利用2b-RAD技术检测基因组区段缺失变异的应用潜力评价*
2018-07-30程陶然李语丽刘平平杨志辉张玲玲胡晓丽包振民
程陶然,李语丽,刘平平,杨志辉,张玲玲,胡晓丽,包振民,王 师
(中国海洋大学海洋生命学院,海洋生物遗传学与育种教育部重点实验室,山东 青岛 266003)
结构变异是个体基因组结构上的微观和亚微观变异,通常包括缺失、重复、插入、拷贝数变异、倒置和易位等,一般结构变异片段的大小介于1 kb到3 Mb之间[1]。从核苷酸的数量来看,结构变异所含的核苷酸总数远远超过SNPs,从而丰富了DNA遗传变异的多样性[2]。缺失是结构变异中一种常见的变异形式,是重要的变异来源之一。缺失变异是同正常染色体相比,个体染色体某一区段丢失,丢失区域大小从单个碱基到整条染色体。导致缺失变异的来源多种多样,如易位丢失,减数分裂过程中染色体交叉错误等,但总的归结为是由不等的互换所导致,如图1所示,由于不等的互换,将序列1上B区段转移至1’序列上,造成序列1上B/B’区段的缺失。
缺失变异按照对个体的影响程度可分为3种:(1)影响微小。缺失片段处于非基因区,对个体的生长发育影响微小。(2)导致疾病,但不影响个体生存。缺失片段处于基因区,造成部分基因丢失,对个体的生长发育造成一定影响,如威廉斯综合征[3-4]。此外,很多遗传疾病也与缺失变异有关,如杜氏肌萎缩症[5]等。(3)造成胚胎或幼苗的致死性危害。缺失片段处于重要的基因区,使个体生存的重要基因丢失,造成个体无法存活,如SMN编码基因的缺失导致脊髓性肌肉萎缩症(Spinal muscular atrophy, SMA),是婴儿死亡的最常见的遗传原因[6]。目前全基因组缺失变异的分析平台(如重测序、SNP芯片等)价格昂贵,难于实现对大量个体的低成本分析。简化基因组测序(Reduced-representation genome sequencing,RRGS)[7]将高通量测序技术[8]和限制性内切酶的使用结合起来,通过对基因组部分位点进行深度测序,大大降低了测序成本。其获取的遗传变异信息目前主要用于SNPs分型,对于结构变异的探索还非常少。简化基因组测序的代表技术有RAD-seq(Restriction-site associated DNA sequencing)[9-10]、2b-RAD(IIb restriction site-associated DNA)[11]、ddRAD-seq(“Double-digest” restriction site-associated DNA sequencing)[12]和GBS(Genotyping-by-sequencing)[13],其中,由于RAD-seq酶切所产生的限制性片段长短不一,影响测序位点的基因组代表性及位点间测序深度的均匀度[14],从而增大结构变异检测的难度。而2b-RAD技术一方面继承了RAD-seq的大部分优点,同时通过改进其所使用的酶(BsaXI)来获得片段长度均一的酶切标签,保证了测序位点的基因组代表性和测序深度的均匀性,从而为结构变异研究提供了可能性。

图1 缺失变异产生的主要来源:不等互换Fig.1 The main generation source of deletion variation: Unequal crossing over
本文选择稳定可靠的2b-RAD测序数据,讨论2b-RAD数据不同测序深度和不同缺失区域大小对检测缺失变异的影响及解决方法,并用拟南芥2b-RAD半模拟数据进行验证。
1 缺失片段鉴定的原理
全基因组测序中已证实基因组中某一序列下读取reads深度可以反映该位点在基因组的情况,如拷贝数变异与reads深度成正比[15],同理该理论可利用每个位点的深度信息推测基因组中缺失区域,即reads深度为零的区域为缺失区域。然而简化基因组测序的酶切标签在基因组间断性分布,导致数据中仅获得部分位点的深度信息。理论上对于某个体基因组的缺失片段,若该片段中含有2b-RAD的酶切标签,在数据比对时,该标签下reads深度为零。所以从比对结果中基因组酶切标签的reads深度出发,若某标签下reads深度为零,说明测序个体中该标签所在的片段可能是缺失的。若相邻的几个标签下reads深度均为零,则认为这些标签所在的片段是缺失片段。但实际情况中,简化基因组数据的测序深度往往对酶切标签深度有较大影响,如某标签下reads深度为零,并不能说明该标签一定是缺失的,而可能由测序错误或测序深度不足导致。
实际数据中可能的缺失情况如图2a所示,当测序后由于测序数据量不足时,可能会出现3种情况:(1)部分非缺失的区段由于没有被测到而形成缺失片段,这种情况在低深度数据中是常见而且不可避免的。(2)部分非缺失片段没有被测到,但这些片段的上游或下游区域存在一个真实的缺失片段,从而导致这个缺失片段的延长。因为从组成上来说,被延长的缺失片段的确含有真实的缺失区间,因此在统计中不会影响缺失片段的检出率,但会对缺失区间大小的判断产生一定的影响。(3)因测序数据量不足导致部分非缺失片段没有reads覆盖,恰好将两个真实的缺失片段连成一个大缺失片段,与情况(2)一样,不会影响缺失片段的检出率,但对正确判断缺失区间的大小带来影响。

(①Genome;②Sequencing data;③Ref tags of genome;④Reads of sequencing data;⑤Real deletion regions;⑥False deletion regions;⑦Data;⑧Reads mapping;⑨Reads depth;⑩Calculate continuous deletion tags;Unrelible decetion regions;Reliable decetion regions.a.基因组中真实缺失区域在测序数据中可能会出现的3种情况;b.检测缺失区间的流程。a.The three cases of the real deletion regions in the genome may appear in the sequencing data; b.Process for detecting deletion regions.)
图2 缺失片段的鉴定原理和处理流程
Fig.2 The identification principle and process flow of deletion regions
综合考虑以上因素,研究中制定的缺失区间检测流程如图2b所示。为了降低缺失片段检测的错误率,对比对结果中所有无reads覆盖的酶切标签进行筛选,当标签中无reads覆盖的连续标签数(continuous deletion tags, cdt)大于某一个阈值cdtset时,认为该处存在缺失变异,反之认为该处是由于外部因素所导致的不可靠的缺失区域,在数据处理过程中cdtset设置为3。为了明确缺失区域的大小,我们人为定义了缺失区域的起始和终止位置,其中起始位置为该缺失区域前最近一个酶切标签的末尾,终止位置是该缺失片段后第一个酶切标签的起始,如图3所示。判断不同深度测序数据对缺失区域检测的影响时,我们用错误率作为评估参数,其计算公式如下:错误率=(检测到的缺失片段数-实际缺失片段数)/检测到的缺失片段数%。

(①Genome;②Restriction digestion tags;③Artifically specificed deletion region size;④Detected deletion region)
图3 缺失片段大小的计算
Fig.3 Calculation of deletion regions’size
2 拟南芥半模拟数据准备
首先利用RADTyping软件[16]中的Extract_cut_site.pl脚本在拟南芥基因组序列(https://www.ncbi.nlm.nih.gov/genome/?term=Arabidopsis%20thaliana)中提取出39 678个BsaXI酶的基因组酶切标签(ref标签)用作后续序列比对的参考序列。
本文采用的拟南芥2b-RAD数据来源于Wang等[17]的5标签串联的2b-RAD实验,且该拟南芥的Multi-isoRAD文库(SRP068382)含有同一个拟南芥个体的5个串联文库,依次命名为AL1-AL5,每个文库的测序深度均在200×左右。随机挑选AL4文库作为测试数据,为了避免文库自身由于测序不足产生的零reads覆盖标签对检测的影响,人为将AL4文库中59个零覆盖标签补齐,标签深度按照文库平均深度的泊松分布随机分配。
对于已不存在零覆盖标签的AL4文库,分别做以下处理:(1)分别构建5个不同大小(5、10、50、100和500 kb)的缺失片段文库。对于每种规格的缺失片段,分别在拟南芥基因组上随机选取50个无重叠的区域(每条染色体选10个),并要求这些区域内至少含有3个或3个以上的ref标签。利用SOAP2软件[18]的比对结果,剔除AL4文库中比对到人为选择的片段含有的ref标签下的reads,分别生成含有真实缺失片段的5个缺失文库。2)在1)所得数据中按一定梯度对测序数据进行抽样。将上述5个缺失文库分别抽取5×-100×生成梯度测序深度数据,并用SOAP2软件与基因组ref标签比对,通过比对结果获取ref标签的reads覆盖深度信息,统计连续无reads覆盖的标签区域。
3 效果评价
本文利用拟南芥半模拟数据分别讨论了不同测序深度和不同大小缺失片段对缺失变异区域检测的准确性的影响。
3.1 测序深度的影响
以片段大小为100 kb的缺失文库的梯度测序数据为例,研究不同测序深度对检测缺失变异准确性的影响。检测到的连续无reads比对的酶切标签区域(即缺失片段)及与根据已知缺失片段计算的检测错误率的统计结果如表1所示。
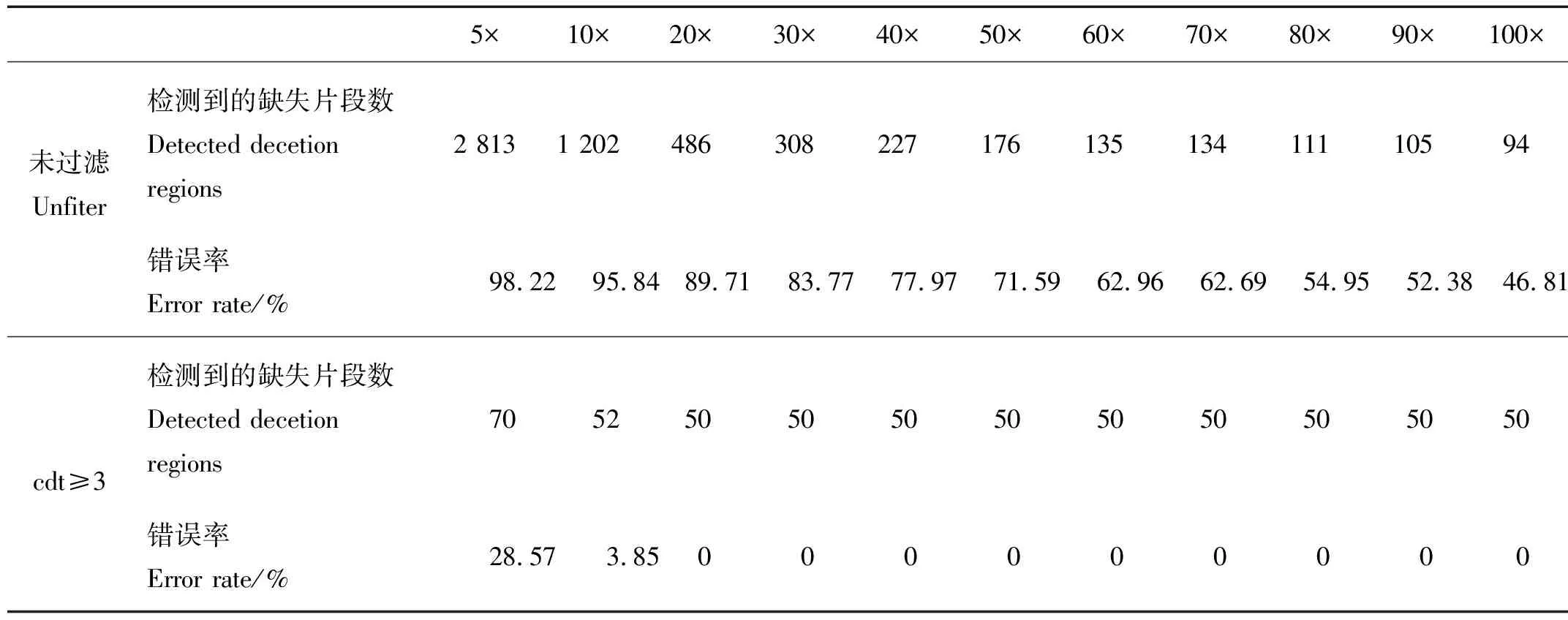
表1 拟南芥100 kb缺失文库的梯度测序数据缺失片段检测的统计结果Table 1 The detection statistics of deletion regions in the gradient sequencing data extracted from A.thaliana 100 kb deletion library
从表1中可以看出,未处理时,测序深度越浅,数据量越少,大部分标签未被测到,导致了较高的缺失片段检测的错误率,如5×、10×时,其错误率高达90%以上。当测序深度增大时,检测到的缺失片段数目逐渐趋近于真实缺失片段数,错误率有所降低,但当测序深度达到100×时错误率仍高达45%以上。这说明直接将连续无reads比对的ref标签所在区域视为缺失区域,会带来很高的错误率。
为了保证检测缺失片段的准确性,对连续无reads比对的ref标签区域进行过滤筛选,要求连续无reads比对的标签数cdt大于某一个阈值cdtset时,认为该处存在的缺失变异是可靠的。当cdtset=3,即cdt≥3时,检测到的缺失片段数接近真实缺失片段数,错误率也随之降低。例如5×测序数据未过滤时检测到2 813个缺失片段,错误率为98.84%,cdt≥3过滤处理后,检测到的片段数目为70,错误率降低至28.57%。表1梯度测序深度下检测缺失片段的结果显示,当测序深度达到20×以上时,cdt≥3条件下的错误率均降到0,说明cdt≥3条件下20×的测序深度即可正确检测测序数据中缺失变异的区间。
理论上真实缺失片段大小约为100 kb,所以梯度测序深度数据统计的缺失片段大小范围应均在100 kb左右。图4-a中显示,当未作任何处理时,造成大量无reads比对的区域被误判成缺失片段,导致箱型图波动较大,尤其是当测序深度过低时,检测到的缺失片段大小的中位值被拉低,远远低于100 kb。当要求连续缺失标签数cdt≥3时,如图4b中所示,除了5×和10×数据的结果偏离100 kb之外,20×及以上的数据的缺失片段大小基本在100 kb左右,表明检测出来的缺失片段基本和真实的缺失片段相符合,从而进一步证实了方法的准确性。
3.2 缺失片段大小的影响
为了研究不同缺失片段大小对检测缺失变异准确性的影响,将5、10、50、100和500 kb的缺失文库的梯度测序数据分别用soap软件进行比对,获取拟南芥基因组ref标签下reads覆盖深度信息,并统计连续无reads比对的标签区间。表2是真实缺失片段5、10、50、100和500 kb在测序深度约为20×左右时,检测到缺失片段的统计情况。未处理时缺失片段检测的错误率随着真实缺失片段大小的增大略有减小,但都在85%以上,而cdt≥3时,检测的缺失片段数均为50,错误率降至0。而图5显示,20×的测序数据cdt≥3条件下检测到的缺失片段大小的分布与标准缺失片段大小基本吻合。

图4 100 kb大小的缺失文库在抽取不同测序深度下处理前、后检测到的缺失片段大小的分布Fig.4 The size distribution of deletion regions detected before treatment and after treatment at different sequencing depth extract by 100 kb deletion data
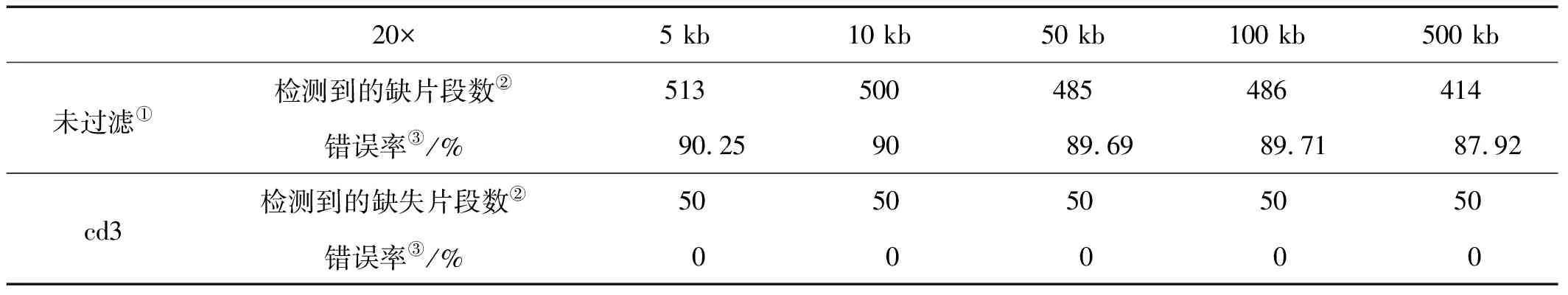
表2 不同缺失片段大小下20×测序数据中检测情况Table 2 The detection summary of deletion regions with different sizes in 20× sequencing data
Note:①Unfitter;②Detected deletion regons;③Error rate
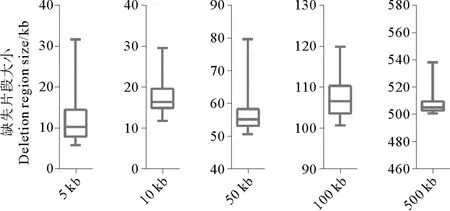
图5 20×测序数据cdt≥3时检测出的缺失片段大小的变化范围Fig.5 Size distribution of detected deletion regions at 20×sequencing depth with cdt≥3
4 讨论
缺失变异是近年来热门的研究之一,但传统的基于SNP芯片和重测序方法的分析平台成本过高,不适于大量个体的缺失变异分析。简化基因组测序将高通量测序技术和限制性内切酶的使用结合起来,通过对基因组部分位点进行深度测序,大大降低了测序成本,而以往简化基因组测序数据主要应用于SNP分型,其中的基因组遗传结构变异等信息没有被充分发掘。
本研究选择了测序深度均一的2b-RAD数据,并克服了简化基因组测序数据中酶切标签在基因组分布上的间断性给缺失变异研究带来的影响。以拟南芥2b-RAD测序数据为研究对象,人为生成包含已知缺失区域的半模拟数据,通过对不同测序深度和不同缺失片段大小的缺失文库分析发现,当测序深度达到20×左右,设定缺失区域所含连续标签数cdt≥3时,即可准确地鉴定测序数据中的缺失区域,且对于不同大小的缺失片段的检测均有效。虽然该方法能大大降低由于抽样造成的假阳性,但是由于我们方法设置的问题,对于小片段(<10 kb)缺失变异的检测仍有一定难度,因此小片段缺失的检测仍需要借助传统方法如重测序等。
本文通过对半模拟数据的分析证实了2b-RAD在缺失变异研究中的可行性,为2b-RAD简化基因组数据检测测序个体中的缺失区域提供了理论指导。在不增加测序成本的条件下,既节约了分析缺失变异的成本,又实现了数据的充分利用,为2b-RAD技术的应用开辟了新方向。
