皮肤型Rosai-Dorfman病14例临床病理分析
2018-07-28樊俊威张丽娟万学峰帕丽达阿布力孜
樊俊威,张丽娟,万学峰,边 毅,帕丽达·阿布力孜
窦状组织细胞增生伴巨大淋巴结病(Rosai-Dorfman's disease,RDD)是一种良性可累及多器官的非朗格汉细胞组织细胞增生症,1969年由Rosai和Dorfman首先报道[1]。RDD通常表现为对称性的无痛性淋巴结增大、发热、多克隆高球蛋白血症、红细胞沉降率升高、免疫功能紊乱等。据估计超过40%的RDD患者有淋巴结外的器官受累,尤其多见累及皮肤[2]。也有报道单纯皮肤型的RDD,但比较少见[3],为此,笔者总结了2012年—2017年新疆医科大学第一附属医院皮肤科收治的皮肤型RDD 14例,并回顾分析其临床资料及相关文献,以提高临床医生对该病的认识。
1 一般资料
复习新疆医科大学第一附属医院2012年1月—2017年6月诊断的所有RDD患者,纳入标准:一般资料填写齐全,经常规伊红-苏木精(HE)染色诊断,并经过免疫组化进一步确诊的皮肤RDD,所有患者均无发热、多克隆高球蛋白血症、红细胞沉降率升高、血常规异常、抗核抗体等免疫学指标异常。排除标准:有系统损害或全身多处淋巴结增加大者,或并发严重系统性疾病者。分析并总结皮肤型RDD的临床组织病理特点,以提高临床医生对该病诊断及治疗的认识。
2 结果
经筛选后共纳入14例经组织病理及免疫组化确诊的皮肤型RDD,其中男5例,女9例 ;汉族12例,维吾尔族1例,回族1例。年龄18~72岁,平均年龄(8±14)岁。病程1个月~2年,平均病程(3.5±2.5)个月。
皮损在单一部位发病者13例,全身多发者1例;同一部位皮损单发者8例,同一部位皮损2处者4例,同一部位皮损3处及以上者各1例。发生于面部者4例,四肢发病者4例,其他部位6例。门诊诊断与组织病理诊断符合并正确诊断为皮肤型RDD者4例,临床误诊为皮肤淋巴细胞浸润者4例,误诊为急性痘疮样苔藓样糠疹者1例,误诊为感染性肉芽肿者2例,诊断为皮损待查者3例。
临床表现为淡红色至棕红色浸润性斑块(图1a)者5例,表现为紫红色至棕红色结节者(图1b)8例,表现为多发的小丘疹者1例;12例患者无明显疼痛及瘙痒等不适症状,1例患者表现为瘙痒,1例患者有疼痛感。
14例RDD组织病理改变示表皮均未见明显异常或轻度萎缩,表皮及真皮之间可见无浸润带;真皮及皮下组织内大量的组织细胞浸润,细胞淡染,可见嗜酸性胞质,细胞核大,呈空泡样,灶性密集淋巴细胞、浆细胞浸润,少量嗜酸粒细胞及中性粒细胞浸润。其中有5例患者皮损的组织病理改变显示部分区域可见组织细胞吞噬淋巴细胞现象(伸入运动)(图2)。
14例确诊为皮肤型RDD患者均完善了免疫组化检查,14例患者均表现为S100及CD68阳性,CD1a阴性(图3)。
14例RDD患者中5例斑块型患者给予皮损内注射复方倍他米松注射液(得宝松)加利多卡因注射液各1 ml,每月1次,随访3个月,2例患者皮损较前明显消退,3例患者效果不明显,加用甲氨蝶呤(MTX)每周7.5 mg,目前仍在随访中;8例结节型患者中7例予以手术切除,随访3个月无复发,1例失访。1例泛发型小丘疹患者,给予口服泼尼松20 mg口服,每个月减量5 mg,沙利度胺50 mg,每日2次口服,随访3个月大部分皮损消退。
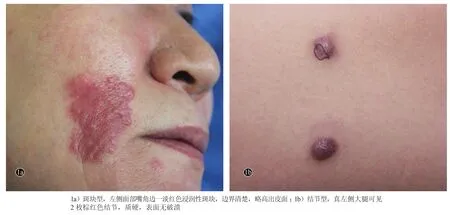
图1 皮肤型Rosai-Dorfman病患者典型临床皮损
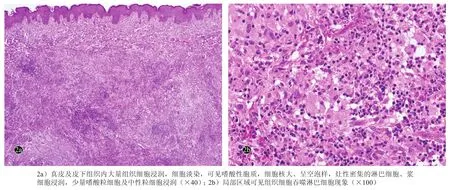
图2 皮肤型Rosai-Dorfman病患者皮损组织病理(HE染色)

图3 皮肤型Rosai-Dorfman病患者皮损免疫组化组织病理(EnVision二步法×40)
3 讨论
RDD病因不明,有人提出假说,可能代表一种细胞介导的免疫异常或是对某种感染(可能是病毒)的过度反应 。尚无EB病毒感染方面的可靠依据 ,人类疱疹病毒(HHV)感染的作用也不明确,在少数患者的淋巴结和皮肤中检测到了人疱疹病毒(HHV)-6迟发相抗原和HHV-6 DNA,血清中检测到抗HHV-6抗体,但目前在聚合酶链反应(PCR)仍未检测到HHV-6主要蛋白衣壳基因。目前该病也没有克隆增殖的依据。
系统性RDD的典型临床表现为无痛性双侧淋巴结增大(常见于颈部和腹股沟),儿童或青年男性较多。尤其在黑人和白种人中更常见,亚洲人口的发病率较低[4]。淋巴结增大往往伴随发热、盗汗、白细胞增多、红细胞沉降率增快及高球蛋白血症(多克隆)。超过40%的患者会发生结外受累,如皮肤、眼眶、中枢神经系统、骨骼,上下呼吸道和泌尿生殖道等[5]。
单纯以皮肤受累而无淋巴结增大的RDD(cutaneous Rosai-Dorfman's disease,CRDD)国内外报道均较少。其典型的皮肤病变是结节、斑块或丘疹,但也有脓疱或痤疮样皮损的报道[6],一般不破溃,也没有明显的临床症状,表现为红色或棕红色的结节或斑块,持续时间为1个月到数年,病变直径可以从数毫米到数厘米。在本组患者中,主要表现为淡红色至棕红色浸润性斑块及紫红色至棕红色结节,也有1例患者表现为多发的小丘疹,临床表现、症状及病程均与国内外报道相同[7]。CRDD发病的年龄较大,女性较多,与经典的RDD相比,CRDD亚洲人的患病率更高[8-10]。本组患者中新疆汉族人发病者较多,可能与就诊人群相关。CRDD患者一般无明显原发疾病及系统受累,无淋巴结增大、发热或盗汗,其实验室检查结果正常, 本组患者均无系统表现。国外报道CRDD好发年龄在50岁左右,本组平均年龄也基本相同。
本组患者均表现为缓慢发生的结节或斑块,病史1个月~2年。皮损颜色暗红色或棕红色,质硬,表面无明显化脓及渗出,患者均无明显临床症状。临床上看到此类患者时应考虑到CRDD,积极完善皮损组织病理检查。组织病理学上RDD表皮通常未见异常或轻度不规则增厚、萎缩,表真皮之间可见无浸润带,真皮内可见弥漫性的组织细胞为主,混杂有淋巴细胞、中性粒细胞、浆细胞的浸润,有时可伴随血管成分增加或间质纤维化。免疫组化显示S-100蛋白和CD68蛋白阳性,CD1a阴性[2]。完整的组织细胞的细胞质中,可以吞噬其他细胞,包括中性粒细胞和淋巴细胞。这一组织病理现象被称为伸入运动,为本病的典型特征。 微观研究表明RDD中的组织细胞缺乏Birbeck颗粒,这是其与朗格汉细胞组织细胞增生症的重要区别[11]。
CRDD的鉴别诊断包括面部肉芽肿、寻常痤疮、鲜红斑痣、玫瑰痤疮、毛发上皮瘤、持久性隆起性红斑、硬化性黏液水肿、汗孔角化症、结节病、硬皮病、麻风、皮肤转移癌等[12]。本组患者中,有4例患者临床诊断正确(28.5%);4例患者误诊为淋巴细胞浸润症,这是由于早期的CRDD临床上也可以表现为境界清楚的水肿性斑块;2例患者误诊为感染性肉芽肿。临床上境界清楚的结节性CRDD也容易与孢子丝菌病、非典型分支杆菌感染等相混肴,后两者往往具有外伤史,表面常常破溃。早期斑块型CRDD与结节型CRDD需要注意与这几种病相鉴别,需进一步行组织病理检查,必要时行病原学培养以明确诊断。
对于CRDD的治疗,虽然16.1%患者皮损可以自行消退,但对皮损持续多年不退者需要积极治疗。治疗的手段包括手术切除、皮损内注射糖皮质激素、口服维A酸、沙利度胺、系统化疗、放疗、激光和冷冻治疗。虽则如此,所有治疗方法的治愈率只有28.6%,其中手术切除为最有效治疗。文献报道80%手术切除的患者经随访可痊愈[13]。本组8例结节型CRDD患者中7例手术切除后痊愈。但对于皮损面积较大的CRDD手术切除治疗容易遗留较大瘢痕。国外有报道使用MTX和小剂量的沙利度胺治疗取得了较好的效果[14,15]。王小坡和孙建方[16]报道口服MTX每周7.5 mg(分3次口服,每次2.5 mg,每次间隔12 h),复方倍他米松注射液(得宝松)1 ml加利多卡因注射液1 ml皮损内适量注射,每月1次,共4次,取得了较好的效果。本组患者中有6例患者给予口服药物治疗,取得了较好的效果,这与国内外报告相同。
目前手术切除治疗被认为是CRDD最有效、最彻底的治疗,所以显得诊断尤其重要,一旦误诊误治,将给患者带来一定的经济损失和肉体上的痛苦[11]。临床上见到皮损为棕红色的斑块,病史较长,皮损质地较硬,组织病理疑似肉芽肿性改变时,要考虑CRDD,要及时行组织病理和免疫组化检查,以明确诊断,减少误诊误治。
