Meleda角化病一家系的SLURP1基因突变研究并文献回顾
2018-07-28林志淼
徐 哲,林志淼
Meleda角化病(mal de Meleda,OMIM 248390)是一种罕见的常染色体隐性遗传的掌跖角化病(palmoplantar keratoderma,PPK),又被称为Meleda病,由Luca Stulli首次发现于地中海东部的现属于克罗地亚的Melada岛。1898年,Neumann首次在文献中报道了本病[1],患病率约1/10万[2]。临床表现为掌跖部位的弥漫角化增生性斑块,手足背侧和肘膝部等四肢伸侧亦可受累。患者生后不久发病,表现为界限分明的掌跖角化伴炎症反应,有异味。常见的特征还包括多汗、口周红斑、关节处苔藓样角化性斑块、锥形指、短指以及指甲肥厚畸形等。其致病基因为位于8q24.3的SLURP1基因[3],编码分泌型尿激酶型纤溶酶原-LY6 /哺乳动物受体相关蛋白1(SLURP1)。SLURP1蛋白是Ly-6蛋白超家族的一个成员,其典型特征是10个半胱氨酸残基之间的二硫键结构以及类似蛇毒素的CCX4CN结构域。SLURP1是α-7-烟碱型乙酰胆碱受体的内源性配体,作为神经调节因子可调节表皮稳态以及肿瘤坏死因子介导的炎症反应[3]。本研究收集到一个Meleda角化病家系,对家系中的成年男性患者、其父母和配偶等人,在获得遗传学研究的知情同意后,进行了基因突变分析。
1 资料与方法
1.1 临床资料

图1 Meleda角化病患者皮损
先证者,男,29岁。因出生时即出现掌跖部位红斑、脱屑,并逐渐扩展加重,累及手足背和肘膝部等,伴随皮肤多汗、皲裂和瘙痒,偶尔出现趾缝间真菌感染而就诊。1岁10月龄时,足背及肘膝部红斑发展为淡红色角化性略隆起的斑块,表面粗糙,有少量角化性鳞屑。手足多汗明显,有腥臭气味,略瘙痒。至3岁龄时,皮损累及到口周、鼻周及臀部,口周和鼻周无明显痛痒等不适。父母体健,非近亲结婚,家族中未见有相似症状的亲属。体格检查:全身各系统体检未见明显异常。皮肤科情况:双侧手掌和足跖对称分布角化性淡黄色斑块,增厚明显,有皲裂伴恶臭,甲板略均匀增厚。皮损延伸至手足背部,跨过掌指(趾)关节直至手足背远端的2/3处,与正常皮肤边界清晰,足缝见白色浸渍(图1a-1c),指(趾)活动不受限。肘、膝伸侧见对称分布的角化性斑块。双侧鼻翼及口周对称性红斑,右侧鼻翼处见刺样凸起(图1d);肛周及会阴部对称性边界清晰的淡黄色角化性斑片,外缘距肛门中心6 cm左右(图1e)。肘部皮损组织病理检查:表皮角化过度,颗粒层增厚,棘层肥厚,可见少量角化不全伴银屑病样增生,真皮乳头延长。真皮上部毛细血管扩张,血管周围稀疏以淋巴细胞为主的炎性浸润(图2)。手足真菌镜检阴性。根据患者自幼出现皮损,进行性加重,表现为累及掌跖及手足背、肘膝部等的角化性斑块,结合组织病理改变,诊断为Meleda角化病。采用5%水杨酸软膏外用2周,脱屑减少,皮损角化增生未见明显好转。
1.2 方法
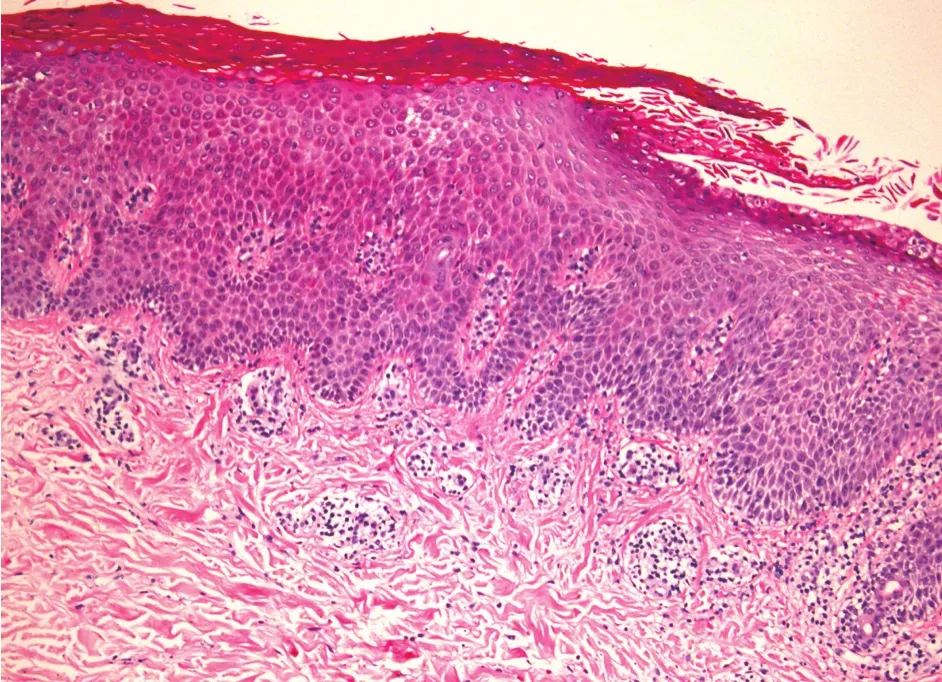
图2 Meleda角化病患者肘部皮损组织病理(HE染色×100)
1.2.1 外周血DNA提取 抽取患者及其父母以及配偶共4人外周血各2 ml,另100例健康人外周血作为对照,血液标本2%EDTA抗凝。患者及其家属签署知情同意书,采用QIAGEN 全血基因组DNA提取试剂盒(QIAGEN,德国),按照说明书标准流程提取基因组DNA。使用NanoDrop2000C超微量分光光度计(Thermo Fisher Scientific,美国)进行DNA浓度及纯度测定。
1.2.2 聚合酶链反应(PCR)引物设计及SLURP1基因的DNA序列扩增 根据GenBank数据库中基因SLURP1的DNA序列(NC_000008.11)及PCR引物设计原则,采用软件Primer Premier 6.0设计特异性引物,共3对。交由上海生工生物工程有限公司合成。引物序列见表1。PCR扩增体系25 μl,反应体系中包括2×LA Taq Premix(Takara,日本)12.5 μl,10 μmol/L上下游引物各1 μl,基因组DNA模板1 μl(50~ 500 ng/μl)。PCR 反应条件为 94 ℃预变性 3 min ;94 ℃ 30 s,退火 30 s,72 ℃ 60 s,30 个循环 ;72 ℃延伸 7 min。
1.2.3 扩增产物测序分析纯化后的PCR产物扩增产物送上海美吉生物医药科技有限公司,采用荧光标记双脱氧末端法在ABI PRISM 3730XL(Applied Biosystems,美国)测序仪上进行测序。应用Chromas软件进行序列读取分析,其结果运用BLAST软件与标准SLURP1序列(NM_020427)进行比对分析。
2 结果
2.1 SLURP1基因突变
在患者SLURP1基因第3外显子检测到纯合突变c.256G>A,该突变导致所编码蛋白的的第86位氨基酸由甘氨酸变为精氨酸(p.G86R)(图3b)。
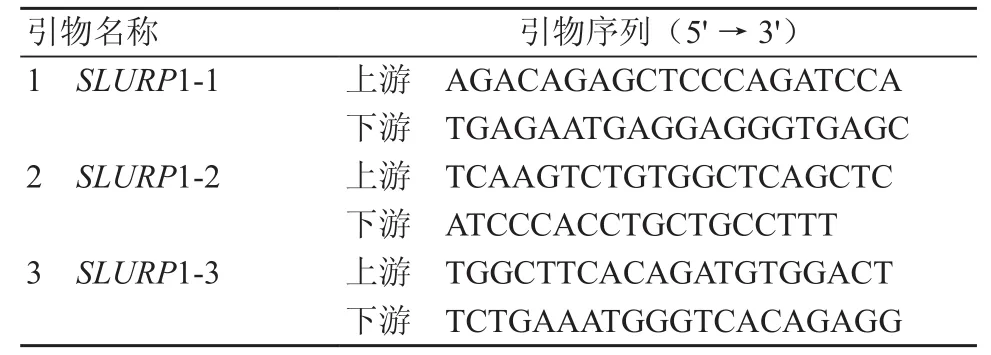
表1 SLURP1引物序列
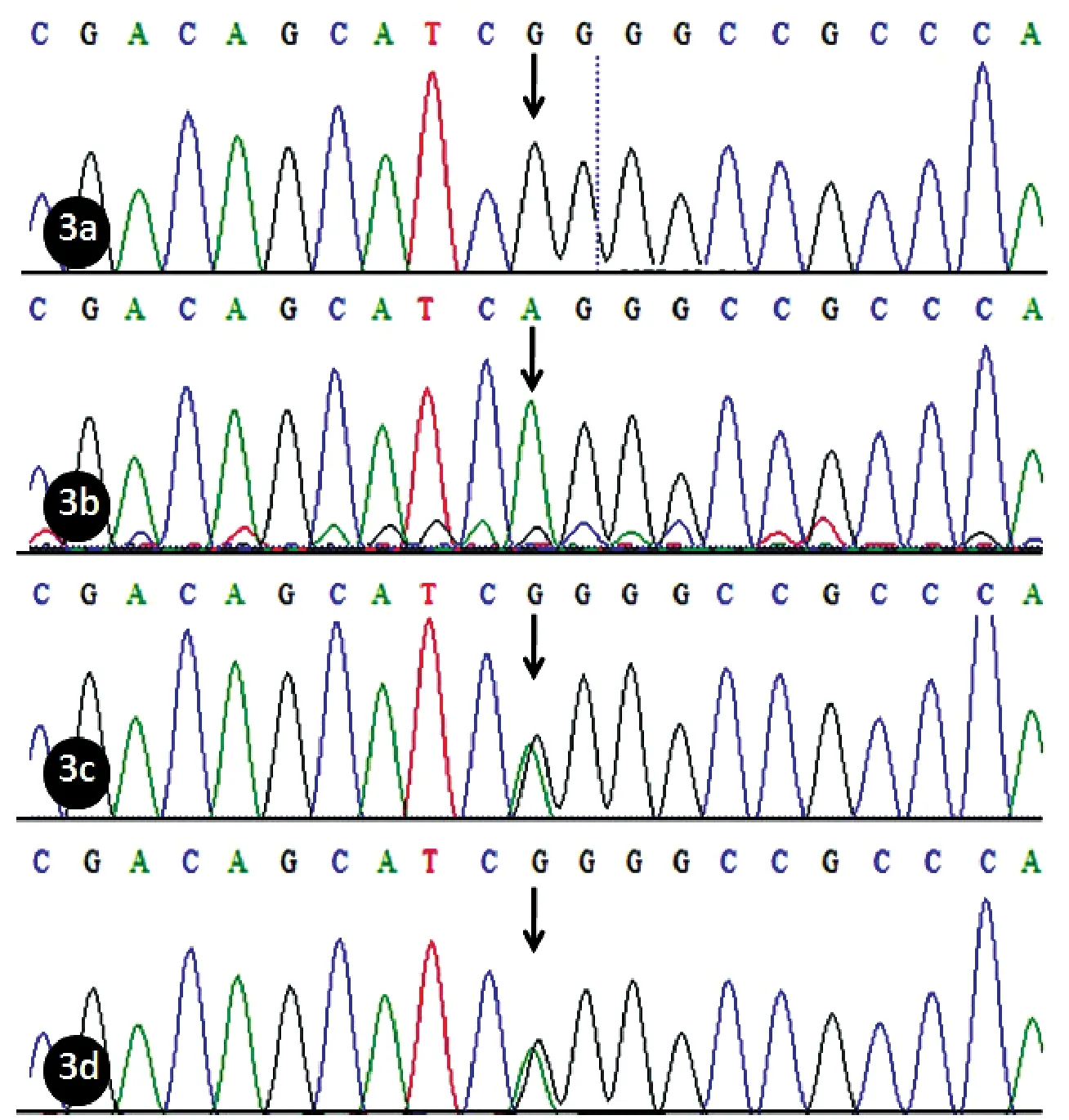
图3 SLURP1基因突变测序图
2.2 突变来源分析
在患者父母检测到SLURP1基因第3外显子的杂合突变c.256G>A(图3c,3d),符合常染色体隐性遗传的致病模式。患者配偶SLURP1基因测序未发现异常。
3 讨论
Meleda角化病是一种罕见的常染色体隐性遗传的掌跖角化病,临床表现为弥漫性掌跖角化增厚,逐渐累及手足背侧及肘膝部。皮肤角化性红斑分布于口周、关节、肛周等处。常见因手足多汗和继发感染引起恶臭以及指(趾)缝浸渍。皮损进展可导致严重的功能障碍,影响手、足活动,甚至出现自发的手足指(趾)假性断指畸形。掌跖皮损其他特征还包括苔藓样病变、短指、甲板畸形和甲营养不良等,该病严重影响患者的生活质量。自从19世纪该病被首次发现以来,至少在19个国家地区相继报道,包括阿尔及利亚,智利,中国,德国,印度,印度尼西亚,意大利,日本,韩国,老挝,利比亚,荷兰,巴基斯坦,阿拉伯,苏格兰,瑞典,突尼斯,土耳其和阿拉伯联合酋长国等[4-9]。本病呈隐性遗传模式,因此具有家族史常常能为诊断提供证据,散发病例相对更少。国内该病少见报道,仅Zhang等[10]首次报道在两个家系中发现3例患儿。王艳等[11]报道1例X连锁鱼鳞病并发Melada角化病患儿,本病例为国内报道的第3例Melada角化病。
Meleda角化病的组织病理表现为表皮弥漫性角质层增生和棘层增厚,但不伴有棘层松解。某些患者可以在组织病理学方面将其作为与表皮松解性鱼鳞病(epidermolyticichthyosis,EI) 等 其他基因相关的角化性疾病的不同之处相鉴别[12]。表皮松解性角化过度是一种组织学模式,可见于一些类型的掌跖角化症、EI或表皮松解性棘皮瘤等皮肤病,主要特点是致密的角化过度,表皮上部棘层松解所致的裂隙或水疱,可见棘层松解细胞等。本病其他的组织病理表现为偶尔可见角化不全及真皮浅层少量炎性细胞浸润等。目前有包括弥漫性PPK、弥漫性残毁性PPK、外胚叶发育不良伴PPK等超过25种掌跖呈角化性皮损的PPK,但需要与Meleda角化病相鉴别的主要是残毁性皮肤角化病(又称Olmsted综合征)和长岛型掌跖角化病[9]。前者呈常染色体显性遗传,临床表现为累及口周及掌跖的红斑和角化,皮损极为严重,在儿童期就可以发生断指等症状,脱发和剧烈瘙痒也是其典型特征;长岛型掌跖角化病主要表现为掌跖部位均匀增厚的红斑角化,伴有遇水后掌跖发白、肿胀和手足多汗,皮损不随年龄增长而加重,通常口周等部位不受累。
Meleda角化病为编码SLURP1蛋白的基因SLURP1突变所致。SLURP1位于8q24.3,由3个外显子组成。SLURP1蛋白是分泌型Ly-6/uPar蛋白超家族中的一员,含有3个结构域,5个双硫键,广泛分布于人体皮肤,尤其是掌跖的角质形成细胞,可以提高烟碱型乙酰胆碱a7受体在表皮角质形成细胞中激活凋亡、诱导分化的作用[13],参与调节角质形成细胞的生长、增殖。当SLURP1基因突变失活时,由于角质形成细胞凋亡异常,导致皮肤高度角化,同时由于SLURP1具有抑制巨噬细胞和角质形成细胞释放肿瘤坏死因子α的功能,SLURP1失活是引起长期炎症反应的诱因[13]。目前,全世界已检测到16个SLURP1基因的突变位点[10],此前中国大陆人群中仅检测到3例患者以及1例X连锁鱼鳞病并发Melada角化病患者的SLURP1突变。其中在两个无关家系均检测到c.256G>A的突变[10],王艳等[11]在X连锁鱼鳞病并发Melada角化病患者检测到c.286C>T的突变,患者均为纯合突变。本研究中患者所检测到的突变为c.256G>A纯合突变,突变来自于杂合的父母携带者,但是父母之间的近亲关系未知。该突变与之前在中国大陆发现的突变一致,p.G86R也曾在韩国,中国台湾以及中东等地区的患者中发现[10,14],提示突变热点的存在。既往认为Meleda角化病在Meleda岛以及中东和地中海地区发病率较高,原因是这些地区,特别是海岛城市中近亲婚配常见,血缘关系较近,提示存在祖先效应。突变所在的第86位甘氨酸位于高度保守结构域,变为精氨酸后可能通过改变蛋白结构,影响表皮稳态以及免疫功能的调节,进而引起患者临床表现。
本研究在一个Meleda角化病家系中检测到成年男性SLURP1基因的纯合错义突变c.256G>A(p.G86R),该突变在国外曾被报道,国内为第2次报道。既往中国大陆首次报道的3例患者均为年龄较小的患儿(2岁、4岁和11岁)[10]。本文患者突变来自于杂合的父母携带者,从基因水平上证实了临床诊断,同时丰富了Meleda角化病的中国患者组成谱。由于患者配偶不携带突变,所以患者所生育的后代为携带者,为其遗传咨询提供了依据。以皮肤角化增生性皮损为主的鱼鳞病、汗孔角化症和PPK等皮肤病,目前主要的治疗方法以润肤、角质剥脱和恢复皮肤屏障功能为主,严重者可以选择口服维A酸类药物[15]。该例患者拒绝选择口服药物治疗,目前治疗仅以外用水杨酸制剂为主,效果不明显。虽然研究显示肿瘤坏死因子α在Meleda角化病炎症方面起着重要作用[9],但是找不到任何关于应用生物或其他免疫调节剂治疗这种掌跖角化性疾病的报道。随着精准医学时代的到来,生物制剂有可能应用于治疗Meleda角化病等角化性疾病。
