婴幼儿配方乳粉中维生素K1的检测
2018-07-11尹丽丽周禹君祝建华刘艳明
尹丽丽,薛 霞,周禹君,郑 红,王 骏,祝建华,刘艳明
(山东省食品药品检验研究院,山东济南 250101)
维生素K1又名叶绿基甲萘醌,是促进血液正常凝固及骨骼生长的重要维生素。维生素K是肝内合成凝血酶原的必需物质,维生素K缺乏时,可导致凝血机制障碍[1]。经常流鼻血者、近期有严重灼伤或外伤者、正服用抗生素者和早产婴儿等都容易发生维生素K缺乏症。在临床上,已将维生素K1用于获得性维生素K依赖性凝血因子缺乏症[2]、新生儿自然出血症[3-4]等维生素K缺乏症的防治。研究表明,在婴幼儿配方乳粉中添加一定量的维生素K1,对于维生素K缺乏症有很好的预防作用。
维生素K1是一种脂溶性维生素,属于多环芳香酮,对热、氧、及水分作用稳定,但在碱性环境下,受阳光照射会分解。
对于维生素K1的检测方法主要有毛细管电泳法[5],分光光度法[6],液相色谱-紫外检测器法[7-11],液相色谱-柱后衍生法[12-14]。其中毛细管电泳法和分光光度法检测步骤繁琐,且精密度不高;高效液相色谱紫外检测器法的灵敏度低;而文献[12-14]中使用高效液相色谱-柱后衍生-荧光检测器时,由于流动相中使用的二氯甲烷对反相色谱系统的脱气包具有强烈的腐蚀作用,对仪器损害较大;此外,国家标准[15]中使用的四氢呋喃对人体危害较大。目前,国家标准[15]中,婴幼儿配方乳粉中维生素K1检测的前处理过程是:用脂肪酶酶解,经碱皂化后,用有机溶剂提取。在此过程中,由于维生素K1在碱性环境中不稳定等原因,往往会导致目标物回收率低,检测结果精密度差。
本文采用高效液相色谱-柱后衍生-荧光检测法对婴幼儿配方乳粉中维生素K1的检测进行了系统性的研究。通过优化流动相条件,减少了对反相色谱系统的仪器损害,提高了方法的灵敏度和选择性;针对方法中的关键步骤-酶解、皂化和萃取进行了分析、总结和优化,使维生素K1的检测方法更加稳定可靠。
1 材料与方法
1.1 材料与仪器
维生素K1标准品 Dr.Ehrenstorfer公司;脂肪酶(酶活力≥700 U/mg) sigma公司;甲醇(色谱纯) 美国Fisher公司;symmetry C18色谱柱(250 mm×4.6 mm,5 μm) 美国Waters公司;锌粉还原柱(50 mm×4.6 mm) 上海安谱实验科技股份有限公司;正己烷(色谱纯) 美国默克公司;酚酞(分析纯) 国药集团化学试剂有限公司;无水乙醇(分析纯) 国药集团化学试剂有限公司;氯化钠(分析纯) 西陇化工股份有限公司;碳酸钾(优级纯) 天津市科密欧化学试剂有限公司;氯化锌(分析纯) 国药集团化学试剂有限公司;乙酸钠(分析纯) 国药集团化学试剂有限公司;婴幼儿配方乳粉 超市购买某知名品牌。
Waters 2695高效液相色谱仪(配荧光检测器) 美国Waters公司;AB204-S型电子天平 瑞士Mettler Toledo公司;SB-800DTD型超声波清洗器 中国宁波新芝生物科技股份有限公司;3-18K型冷冻离心机 德国Sigma公司;mini-Q超纯水制备器 美国MILLIPORE公司;HGC-24型氮吹仪 中国HENGAO公司;恒温水浴振荡器 德国优博莱公司;涡旋混合器 德国IKA公司。
1.2 实验方法
1.2.1 标准溶液的配制
1.2.1.1 标准品溶液的校正 称取维生素K1标准品20.0 mg于10.00 mL容量瓶中,用正己烷溶解并定容,配制成维生素K1母液。取200 μL维生素K1母液至25 mL容量瓶中,用正己烷定容,配制成浓度约为16 μg/mL的校正溶液,用紫外可见分光光度计在248 nm条件下校正(校正方法见[15])。
1.2.1.2 标准工作液的配制 精确移取经校正过的维生素K1储备液,氮气吹干后,用甲醇溶解,配制成浓度为:0.2,0.5,1,2,5 μg/mL的标准工作液。
1.2.2 样品处理过程 准确称取乳粉试样2.5 g(精确到0.01 g),于50 mL离心管中,加入一定量的脂肪酶后,加入10 mL(37±2) ℃温水溶解,涡旋2~3 min后,置于(37±2) ℃恒温水浴振荡器中振荡一定时间,使其充分酶解。取出酶解好的试样,加入10 mL无水乙醇,加入一定量的碱,充分混合均匀,进行皂化反应。加入15 mL正己烷,涡旋5 min,进行萃取。5000 r/min离心3 min,取上清液至50 mL离心管,加入10 mL饱和氯化钠溶液和两滴0.5%酚酞乙醇溶液,涡旋5 min,进行水洗。6000 r/min离心3 min,取上清液至50 mL离心管中,氮气吹干,准确加入2.00 mL甲醇复溶,过有机滤膜后,高效液相色谱仪进行测定。
1.2.3 高效液相色谱条件 symmetry C18色谱柱:250 mm×4.6 mm,5 μm;锌还原柱:50 mm×4.6 mm;流速:1 mL/min;检测波长:激发波长为243 nm,发射波长为430 nm;进样量:10 μL;流动相:甲醇(含有冰乙酸0.03%,氯化锌1.5 g/L,无水乙酸钠0.5 g/L)。
1.2.4 方法学考察
1.2.4.1 线性范围与检出限 将1.2.1.2配制的系列标准工作溶液进行液相色谱条件下测定,以维生素K1峰面积为纵坐标,以标准工作溶液的浓度为横坐标,进行标准曲线的绘制。加标空白样品基质按照1.2.2样品前处理过程,在1.2.3液相色谱条件下进行测定,通过目标物的响应与背景噪音的比值计算维生素K1的方法检出限。
1.2.4.2 精密度实验 对32、72、115μg/100 g三个不同含量的样品分别进行6次独立测定,计算精密度。
1.2.4.3 回收率实验 根据GB/T 27404-2008《实验室质量控制规范 食品理化检验》的要求,结合实际检测样品中维生素K1的含量,对空白样品进行5、10、100 μg/100 g三个水平的加标测定,计算加标样品测定值与样品测定值之差,通过其差值与实际添加的目标物含量之间的比值计算回收率。
1.3 数据处理
通过与仪器配套的Empower 3工作站软件完成数据采集与处理。
2 结果与分析
本文对国家标准[15]中的关键技术-酶解条件、皂化条件和萃取条件和流动相条件进行研究,经过优化并改进,维生素K1的检测方法更加稳定、可靠。
2.1 酶解条件的选择
前处理过程中,首先用脂肪酶降解试样中的不饱和脂肪酸,在酶解过程中,酶的用量对目标物的提取有一定影响,本文比较了酶解时间和脂肪酶用量不同时,目标物的回收率与精密度。由表1可知,酶解6 h和12 h(脂肪酶量为0.8 g条件下),对于目标物含量的测定,效果相当,但是由于前处理操作复杂,包括酶解、皂化、萃取、水洗等步骤,如果样品量少,可选择当天酶解6 h,如果样品量多,当天酶解时间紧迫,建议选择酶解过夜12 h;另一方面,不同脂肪酶量在酶解12 h的条件下,回收率无明显差异,但是酶量越小(即0.2 g和0.4 g酶量)时,后续的萃取步骤中液液两相分层越不明显,结果的精密度越差,当酶量较大(即0.8 g脂肪酶),后续的萃取步骤中液液两相分层明显,结果精密度好,故采用0.8 g脂肪酶酶解12 h的酶解条件。

表1 酶解条件的比较Table 1 Comparison of enzyme solution conditions
2.2 皂化条件的选择
皂化反应中,碱的作用是使酶解后的脂肪生成脂肪酸盐,溶解于水相中,加入量的多少会直接影响回收率。加入量过少会使皂化反应不完全,萃取时不容易分层;过多则会和目标物维生素K1反应,使最后的结果偏低;乙醇的作用是保护维生素K1不参与反应,同时起到消泡促进分层的作用。本文对皂化条件中碱的种类及用量进行了考察,具体结果见表2。由表2可知,皂化用碱若选择NaOH溶液(10 moL/L,2 mL)回收率低,精密度差,若选择NaOH溶液(10 moL/L,0.5 mL)时,精密度差。原因可能是由于两种NaOH溶液碱性太高,目标物与碱反应而被分解,故在此条件下不稳定存在;本文在皂化反应中使用K2CO3固体,碱性相对低,目标物在此条件下稳定存在,因此具有较高的回收率和精密度。

表2 皂化条件的比较Table 2 Comparison of saponification conditions
2.3 萃取条件和水洗条件的比较
萃取是为把目标物维生素K1从皂化后溶液中萃取出来,也是进一步净化的过程。维生素K1是脂溶性维生素,故采用有机溶剂正己烷作为萃取溶剂。文章对于萃取次数进行了考察,发现萃取两次和萃取一次的测定结果基本一致,因此,从提高前处理效率的角度,建议萃取一次。
由于维生素K1对碱液不稳定,所以需要用水把萃取溶剂正己烷中可能携带的碱液洗掉。另外,水洗时加入饱和氯化钠,可防止液液分配过程的乳化。从表3中可以看出,增加水洗步骤,可明显提高目标物含量随时间变化的稳定性,但是水洗2次,目标物含量损失变大,因此,建议水洗一次。
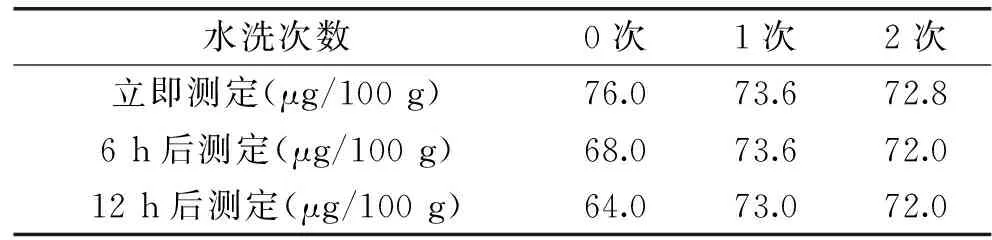
表3 水洗对目标物含量稳定性考察Table 3 The stability of target content by washing
为了使有机相和水相分层明显,保证实验可重复性,水洗过程中需加入两滴酚酞溶液(0.5%)。加入酚酞是否影响目标物含量的测定结果见表4。从表4中可以看出,加入酚酞不影响对目标物含量的测定。

表4 酚酞对目标物的影响Table 4 The effect of phenolphthalein on target objects
2.4 流动相条件的比较
目前,高效液相色谱-柱后衍生-荧光检测的检测方法中通常流动相含有四氢呋喃或二氯甲烷。国家标准[15]中流动相使用的四氢呋喃,四氢呋喃对身体危害较大;文献中[12-14]使用的流动相含有二氯甲烷,二氯甲烷对反相色谱系统的脱气包具有强烈的腐蚀性,长期大量使用会造成脱气包渗漏。本文比较了流动相中是否含有二氯甲烷对目标物分离效果的影响,具体谱图见图1~图4。图1和图2为使用流动相A(甲醇中含有冰乙酸0.03%,氯化锌1.5 g/L,无水乙酸钠0.5 g/L)条件下采集的色谱图。图3和图4为使用流动相B(甲醇中含有二氯甲烷10%,冰乙酸0.03%,氯化锌1.5 g/L,无水乙酸钠0.5 g/L)条件下采集的色谱图。通过比较图2和图4发现,使用流动相A(不含二氯甲烷溶液)进行色谱分析,峰型理想,分离效果相对更好。流动相中不含有二氯甲烷,减少了对仪器反相系统的损害,也减少了对身体的危害,故本文使用流动相条件A。
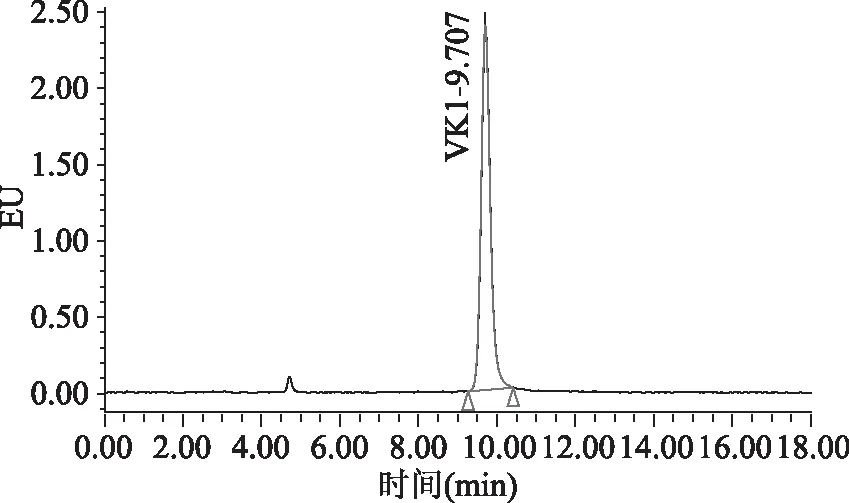
图1 使用流动相A条件下采集的标准品色谱图Fig.1 The chromatogram of standard solution by using phase A
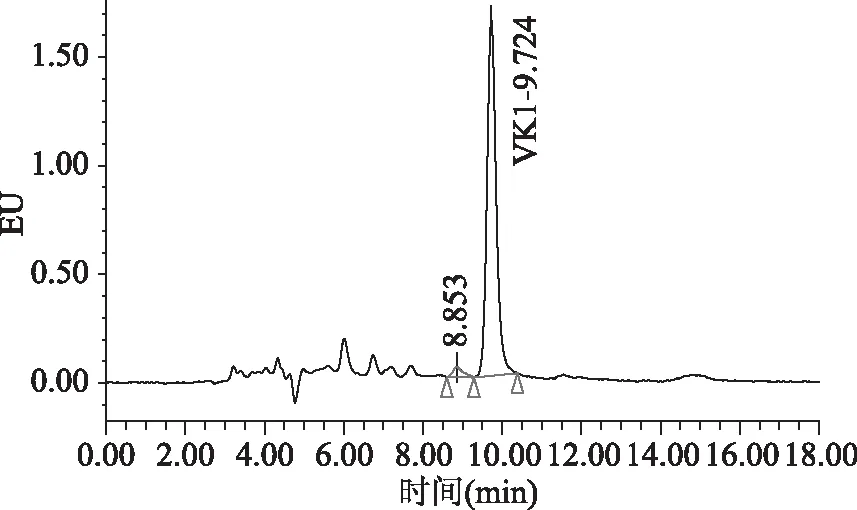
图2 使用流动相A条件下采集的样品的色谱图Fig.2 The chromatogram of samples by using phase A
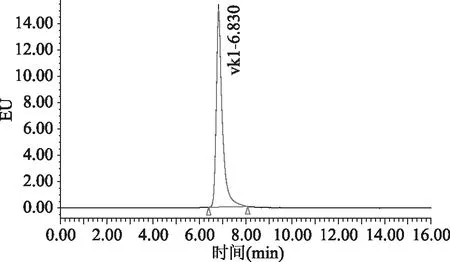
图3 使用流动相B条件下采集的标准品的色谱图Fig.3 The chromatogram of standard solution by using phase B
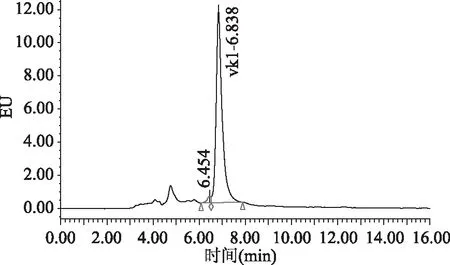
图4 使用流动相B条件下采集的样品的色谱图Fig.4 The chromatogram of samples by using phase B
2.5 方法学考察
2.5.1 线性范围与检出限 标准工作溶液线性回归方程为:Y=2.1E+6X-69808,线性相关系数R2=0.9995。本文采用加标空白样品基质中标样对应的响应与背景噪音的比值(S/N=3计算得到维生素 K1的方法检出限为1 μg/100 g。
2.5.2 精密度实验 由表5可见,三个样品的精密度均小于5%,实验数据稳定,精密度较高。

表5 精密度实验结果Table 5 Results of precision experiment
2.5.3 回收率实验 由表6可知,空白样品进行三个水平的加标测定,回收率在90%~110%之间。本方法对乳粉质控样品NIST SRM 1849a样品进行检测,测定结果为0.97 mg/kg,在标定值(1.06±0.17 mg/kg)的偏差范围内。
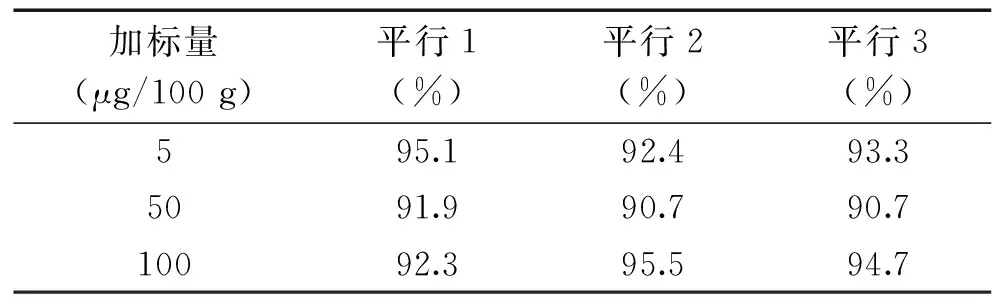
表6 回收率实验结果Table 6 Results of recovery
3 结论
上述实验表明,影响乳粉中维生素K1测定的关键因素有:酶解条件、皂化条件、萃取条件和流动相条件。本文通过对关键因素的考察,确定了以下实验条件,包括酶解条件:酶解量0.8 g,酶解时间12 h;皂化条件:碳酸钾1.0 g;萃取条件:正己烷萃取一次,饱和氯化钠水洗一次;流动相条件:甲醇(含有冰乙酸0.03%,氯化锌1.5 g/L,无水乙酸钠0.5 g/L)。
综上所述,本文通过对方法中关键技术条件的优化,使维生素K1的检测方法操作简单、高效、准确度高,同时减少了对液相色谱仪-反相系统的损伤,降低了对检验人员身体的危害。本方法不仅可以为生产企业控制维生素K1的添加量、准确标示给予指导,还可以为科研机构积累、总结维生素K1含量范围及趋势提供技术支持。
