EZH1/2在心血管发育及其疾病中的研究进展
2018-07-10陈瑞平陈寄梅郭惠明
杨 斌,陈瑞平,杨 雷,罗 玲,庄 建,陈寄梅,郭惠明,朱 平
(1南方医科大学第二临床医学院,广东 广州510515;2广东省心血管病研究所,广东省人民医院,广东省医学科学院,广东 广州510080;3华南理工大学医学院,广东 广州 510006)
0 引言
目前对心血管系统发育及其疾病的研究在基因表达及信号通路相关的分子机制方面已经取得了很多突破,主要涉及发育、疾病相关的转录因子[包括GATA 家族、NKX 家族、MEF(myocyte enhancer fac⁃tor) 家族、Tbx(T⁃box DNA⁃binding protein) 家族和HAND(the heart and neural crest derivatives expressed transcript genes)家族等]、致病基因、非编码RNA、表观修饰因子及其复合物等[1]。
多梳蛋白家族(polycomb group,PcG)是一组通过修饰染色质来抑制靶基因转录的表观调控因子,从果蝇到哺乳动物存在高度的保守性;多梳蛋白抑制性复合体2(polycomb repressive complex 2,PRC2)是该家族中最重要的蛋白复合体之一,它包含四个核心亚基,分别为zeste基因增强子人类同源物1/2(enhancer of zeste homolog 1/2,EZH1/2)、zeste 基因抑制子 12(suppressor of zeste 12,SUZ12)、胚胎外胚层发育蛋白(embryonic ectoderm development,EED)以及视网膜母细胞瘤结合蛋白 7/4(retinoblastoma binding pro⁃tein 7/4,RBBP7/4)[2]。 其中,EZH1/2 作为 PRC2复合物的催化亚基,具有催化组蛋白H3第27或9位赖氨酸三甲基化的甲基转移酶活性,是重要的转录抑制因子,参与染色质结构变化、基因表达以及生长调控等过程。
EZH1/2通过参与心脏发育和心血管疾病发生发展过程中的特异性调控网络,在心脏发育缺陷、动脉内皮损伤、斑块形成、心室重构、心力衰竭以及心肌再生与修复中存在典型的表达及活性差异,进一步影响其病理生理进程。
1 EZH1/2的结构和功能
EZH2是果蝇zeste基因增强子的人类同源物,位于人类染色体7q35[3],包含20个外显子和19个内含子,编码由746个氨基酸残基构成的蛋白分子。其N端有一个WD重复结构域,该结构域与EED及SUZ12的靶定有关;C端则包含一个与组蛋白甲基转移酶活性相关的SET结构域[4]。EZH1和EZH2互为同源物,具有相同的作用靶点,并且两者的表达在时间和空间上具有互补性,而在心脏发育早期,EZH2发挥主要的生物学功能[5]。EZH1/2是PRC2的核心亚基,具有组蛋白甲基转移酶活性,主要参与核小体组蛋白H3第27位氨基酸残基的三甲基化修饰(H3K27me3),进而招募 PRC1并引起组蛋白H2AK119的单泛素化,促使染色质凝结,阻止RNA聚合酶Ⅱ依赖的转录延伸,最终抑制靶基因转录。此外,EZH2还可以与组蛋白去乙酰化酶1/2(histone deacetylase1/2,HDAC1/2)及 DNA 甲基转移酶(DNA methyltransferase,DNMT)协同作用从而强化基因转录抑制效应,或直接抑制基因转录(非经典通路/不招募 PRC1)[6-7]。 EZH1/2 协同 PcG 蛋白家族的其他组分及多个共作用因子,通过抑制心脏发育中的关键基因及转录因子的转录等途径,在心脏发育和心血管疾病发生发展过程中起着重要的调控作用。
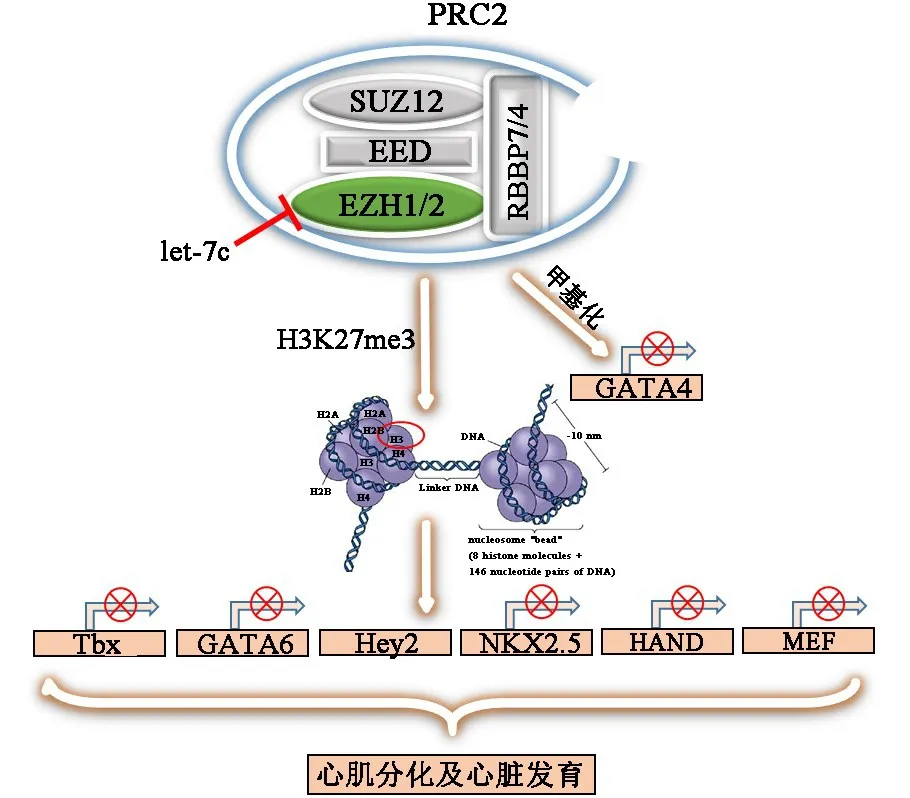
图1 EZH1/2(PRC2)对心肌分化及心脏发育的调控
2 EZH1/2在心脏发育中的作用
心脏发育是一个极其复杂的过程。一方面,心脏发育需要多个转录因子及信号通路精确调控相关基因在时间和空间的精确表达,另一方面,转录因子及信号通路本身也受到表观修饰等作用的调控。人类心脏发育需经历以下几个阶段:①来源于中胚层的心脏前体细胞从原始条纹处迁移至生心板对称的心嵴,经分化向中线扩增形成新月形的细胞嵴;②细胞嵴在中线处吻合形成原始心管;③心管延长并向右弯曲、环化,逐步形成房室腔、房室分隔、瓣膜、流出道结构、心脏传导系统及冠状脉管循环等,最终发育为完整的心脏[8]。其中,与心脏发育密切相关的基因有ISL1(ISL LIM homeobox 1)、ACFC1、DNAH 及 JAG;与其相关的转录因子/转录因子家族有 GATA、MEF、NKX2.5、Tbx、HAND 等;同时参与心脏发育的信号通路包括 NODAL、NOTCH、WNT、BMP 等[1,9]。
在心脏发育中,EZH2通过表观组蛋白修饰来沉默心脏转录因子的表达,对早期胚胎干细胞向心脏细胞系的分化及心脏基因的表达具有重要的调控作用,并参与婴儿心脏内稳态 的 维持[5,10]。 研究[10-12]表明,在心脏发育和心肌细胞分化过程中,EZH2与PcG家族及非PcG家族中的部分成员[包括长链非编码RNA(lncRNA)以及microRNA等]协同发挥表观调控作用。已经确定有50多个基因在心脏发育中受EZH2及PcG家族成员的协同调控,其中包括Isl1、Tbx2、Tbx3、AND1、Irx5(Iroquois homeobox 5)和Six1[12]。
进一步研究[13]表明,EZH2主要调控心脏前体细胞的增殖及分化,而EZH1主要负责维持围产期婴儿心肌细胞的增殖以及成人心梗后心肌细胞的增殖和再生,EZH1和EZH2对心肌细胞的增殖与分化及胚胎发育晚期心肌细胞成熟的调控方面存在一定的重叠作用。EZH2基因敲除的小鼠胚胎在原肠胚未形成之前胚胎就发生死亡[12,14]。而通过组织特异性敲除EZH2基因后,小鼠会出现致密性心肌发育不全、过度小梁化和心室中隔缺损等先天性心脏畸形。另有研究[15]发现,Hey2是EZH2的下游靶点基因,在心肌细胞增殖及心脏形态形成过程中具有重要的作用,并且EZH2对Hey2的调控是独立于Notch信号活性的。相应地,敲除EZH1的胚胎发育正常,但会影响出生后心肌梗塞的心肌再生及心脏功能[15]。
研究发现,EZH2是let⁃7c的下游直接靶位点,过表达let⁃7c可以促进中胚层心脏特异细胞系的表达,这是通过let⁃7c靶向EZH2进一步抑制其转录及活性实现的。此外,let⁃7c可以调控心脏转录因子(如Nkx2.5、Mef2c、Tbx5)启动子区域组蛋白的甲基化修饰,从而促进心脏分化。在过表达let⁃7c的细胞中,发现心脏转录因子启动子区域的抑制性修饰标记(H3K27me3)降低,同时发现激活性修饰标记(H3K4me3)上调,这说明EZH2及其组蛋白修饰作用在心脏分化过程中是必不可少的[10]。
EZH2不仅参与组蛋白的甲基化修饰,也调控非组蛋白的甲基化修饰,如EZH2可以使心脏转录因子GATA4的第299位赖氨酸甲基化,从而抑制其转录。作为人及小鼠胎儿心脏发育中的关键剂量依赖转录调控因子,GATA4可以将p300招募至特定的染色质位点,而p300乙酰化GATA4可以激活GATA4的转录活性。在胎儿心脏中,EZH2通过甲基化修饰GA⁃TA4的第299位赖氨酸来抑制GATA4对p300的招募,进而抑制GATA4的转录活性及基因表达。但是,过度抑制会阻碍胚胎干细胞向心肌细胞的分化,导致心脏发育不完全;相反地,GATA4的过度转录又会引起心肌肥大,所以EZH2直接抑制GATA4的不正常转录可以防止心肌的过度增生[16]。
3 EZH1/2在主动脉夹层及先天性心脏病中的作用
3.1 主动脉夹层及二叶型主动脉瓣 人全基因组DNA甲基化检测结果显示:在升主动脉夹层(ascending aortic dissection,AD)及二叶型主动脉瓣(bicuspid aortic valve,BAV)患者中存在非CpG位点(包括 CpA、CpT 和 CpC)的甲基化缺失[17]。 与其他位点不同,CpG位点在AD和BAV中的甲基化存在差异。BAV中存在H3K27me3的大量富集,表现为EZH2靶点的超甲基化及EZH2酶活性增加;而DNA甲基化基因表达分析结果显示:在AD中主要表现为平滑肌细胞的去分化,这说明EZH2在BAV和AD的发生发展中可能起到了重要的调控作用[17]。

图2 EZH2在DiGeorge综合征发展中的作用
3.2 DiGeorge/Velocardiofacial综合征 DiGeorge/Velocardiofacial综合征又叫做22q11.2微缺失综合征,主要由Tbx1染色质基因丢失引起,Tbx1促进第二心区细胞的增殖并抑制其分化,这些细胞主要迁移并富集形成流出道及右心室结构,Tbx1功能缺失或者表达不足可以导致心脏流出道发育不全,进而出现主动脉弓及心脏流出道畸形等类似于DiGeorge/Ve⁃locardiofacial综合征的疾病表型。研究[18]表明,Tbx1与p53通过占据Gbx2基因上的相同靶点元件而产生强烈的竞争作用。而Trp53基因敲除或者药物抑制后可以明显修复Tbx1杂合型基因突变造成的心血管缺陷。进一步研究[18]发现,p53可以间接招募EZH2并正向调节EZH2和H3K27me3水平,使其富集在Tbx1和p53共同占据的基因元件上。因此,抑制p53的表达,可以改变p53和Tbx1共同占据的Gbx2基因元件的染色质状态,使其H3K27me3水平显著降低,从而促进Tbx1对Gbx2基因的调节作用。
3.3 其他心血管畸形 在小鼠胎心中,PRC2的三个主要亚基(Eed、EZH2和 Suz12)的 mRNA水平及蛋白水平表达显著。而在成年心脏中,三个亚基的转录水平都明显下调,甚至检测不到EZH2和Eed的表达蛋白[19]。EZH2可以通过抑制 Ink4a/b的表达来促进心脏增殖[19]。在心脏发育早期一个很窄的窗口期,对心脏进行特异的条件性敲除EZH2可引起心脏非特异基因的表达,导致心脏后续发育紊乱甚至形成致死性心脏畸形,包括心肌发育不全、过度小梁化、室间隔缺损等。相反地,条件性敲除发育后期心脏(晚期胚胎/分化的心肌细胞)的EZH2基因则不影响心脏的正常发育[19]。类似的研究也得出相似的结论:EZH2在房室分割以及正常的流出道形成过程中是非常重要的。在胚胎发育早期的心脏前体细胞中条件性敲除(Nkx2.5⁃Cre)EZH2,发现小鼠胚胎心脏可因胚胎干细胞发育缺陷而出现心内膜垫发育不全,并抑制内膜向间充质转化,抑制心肌增殖并促进细胞坏死,导致心脏发育畸形,包括右室双流出道、永存动脉干、膜性/肌性室间隔缺损、房间隔缺损、房室管畸形等,最终导致小鼠于围产期死亡或出生后发生心肌纤维化[15]。 另有研究[20]发现,在人的胚胎干细胞中,EZH2被NR2F2招募到OCT4上,可以抑制OCT4在心肌分化过程中的表达,NR2F2基因发生突变会引起心脏房间隔缺损。
另有研究[21]表明,组蛋白去乙酰化酶3(histone deacetylase 3,HDAC3)协同转化生长因子 β1(trans⁃forming growth factor⁃β1,TGF⁃β1)去乙酰化非依赖的表观沉默作用,调控第二生心区的发育。约2/3的人类先天性心脏疾病与第二生心区来源的结构改变有
关,因此,第二生心区的表观调节异常是先天性心脏疾病的诱发因素[21]。 TGF⁃β1通过非依赖去乙酰化途径来调节第二生心区心脏结构的发育[21]。小鼠胚胎第二生心区缺乏HDAC3会促进TGF⁃β1的生物利用,使内皮间质转化发生异常并改变细胞外基质的稳态,从而引起升主动脉扩张、流出道畸形、主动脉骑跨、右心室双出口、半月瓣膜发育异常、二叶型主动脉瓣、室间隔缺损等先天性心脏病甚至发生胚胎死亡。而药物抑制 TGF⁃β 可以修复这些缺陷[21]。 研究[21]表明,EZH2、EED和 SUZ12同时被 HDAC3招募到NCOR复合物上,来促进 TGF⁃β1调控区的H3K27me3修饰,从而维持TGF⁃β1在第二心区间质的沉默。
3.4 唐氏综合征伴随先天性心脏发育缺陷 唐氏综合征(down syndrome,DS)是一种最常见的遗传性智力缺陷综合征,由21号染色体发生三体、易位或嵌合引起,常伴有先天性心脏发育缺陷等疾病。相比正常的小鼠,DS小鼠模型海马中(Ts65Dn mouse model)存在一系列的miRNAs显著上调或下调,这些miR⁃NAs的潜在靶点中,Ship1、Mecp2和 EZH2都在 mR⁃NA水平存在显著下调[22]。

图3 EZH2在血管发育中的作用
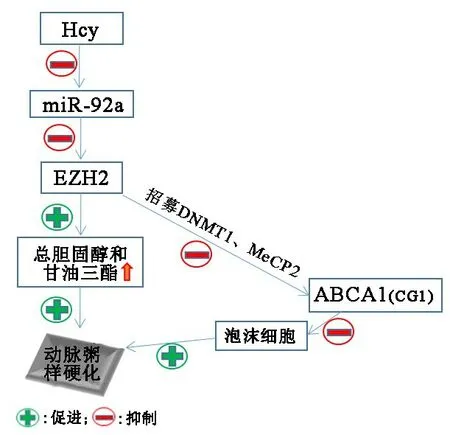
图4 EZH2在动脉粥样硬化发展中的作用
4 EZH2/1在动脉瘤及动脉粥样硬化中的作用
许多心血管疾病的病理进程都伴随有基质金属蛋白酶(matrix metalloproteinases,MMPs)活性的增加,如主动脉瘤中血管发育不完整、动脉粥样硬化斑块破裂等。MMP9是细胞外基质在发育和疾病中保持机体内平衡的重要调控因素。研究[23]发现,EZH2可以靶向MMPs及其上游转录激活因子Fosl1、klf5和Creb3l1(Creb不仅可以直接激活内皮细胞MMP9的启动子,诱导内源性的MMP9表达,还可以增强动脉粥样硬化中的炎症反应),通过抑制MMPs及其激活因子的表达来维持血管发育的完整性。在缺乏EZH2的小鼠胚胎中,因为去除了对MMP9的抑制,可引起脉管系统发育不完整,而MMP9基因被抑制后可以修复血管发育的不完整性。在人脐静脉内皮细胞中,EZH2还可以通过调节细胞的粘附及交流来促进血管生成。也有研究[24]发现MMP9在癌症及炎症环境中表达急剧上调,EZH2作为MMP9的重要抑制因子,可以为癌症及脉管系统疾病提供新的治疗策略。但因为EZH2过表达与许多癌症的侵袭性及预后相关,具体机制不详,所以,EZH2作为治疗靶点的同时也需要考虑到其副作用。
高同型半胱氨酸血症(hyperhomocysteinemia,HHcy)是心血管疾病的独立危险因素,尤其是动脉粥样硬化。 研究[25]发现,Hcy(homocysteinemia)诱导的动脉粥样硬化是由miR⁃92a调控的EZH2表达上调实现的。在敲除ApoE的小鼠模型中,以高蛋氨酸喂食16周后,发现EZH2和H3K27me3水平均上调,同时miR⁃92a表示下调。已经明确过表达EZH2可提高泡沫细胞中H3K27me3水平,促进总胆固醇和甘油三酯的堆积,而在泡沫细胞中上调miR⁃92a可以减少EZH2的表达。因此,EZH2在同型半胱氨酸调控的脂代谢异常中发挥了关键作用,miR⁃92a可以成为一个新的同型半胱氨酸相关的动脉粥样硬化的治疗靶点[25]。 也有研究[26]表明,三磷酸腺苷结合盒转运子A1(ATP⁃binding cassette transporter A1,ABCA1)在维持细胞胆固醇体内平衡中具有重要作用,而ABCA1表达的表观修饰影响巨噬细胞源性泡沫细胞的形成及动脉粥样硬化的发展。ABCA1启动子区含有大量的CpG 岛(CpGisland,CGI),EZH2诱导 DNA甲基转移酶1(DNMT1)的表达,招募CpG岛甲基结合蛋白2(methyl⁃CpG⁃binding protein⁃2,MeCP2),并促进 DN⁃MT1、MeCP2与ABCA1的结合。综上,EZH2通过在ABCA1的启动子区进行甲基化修饰抑制ABCA1表达,进而促进泡沫细胞形成并促进动脉粥样硬化的发展。
进一步研究[27]发现,在动脉粥样硬化斑块的平滑肌细胞和炎症性细胞的表观修饰中,H3K9和H3K27的甲基化水平明显降低而乙酰化水平升高,并且与动脉粥样硬化斑块的严重程度有很强的相关性。经免疫组织化学法分析证实,H3K27Me3总体水平的减少与动脉粥样硬化的严重程度成正相关[28]。这说明上述两种组蛋白表观修饰在动脉粥样硬化中起到了决定性的作用。
其次,低密度脂蛋白(low density lipoprotein,LDL)胆固醇是一个可修饰的冠心病危险因子,可以诱导内皮功能失调。而内皮细胞 Kruppel⁃like Factor 2(KLF2)是内皮依赖性血管稳态的重要转录因子[29];KLF2的转录主要由近端启动子调控,该区域富含CpG二核苷酸以及KLF2转录相关的主要调控原件,包括MEF2(上调表达)以及p53(抑制表达)结合位点,而组蛋白修饰因子(包括 EZH2)在调控KLF2 表达中扮演了重要角色[30-31]。
LDL通过诱导内皮DNA甲基转移酶I的表达并促进其酶活性来下调内皮KLF2的表达,敲除DNA甲基转移酶I或使用DNA甲基转移酶抑制剂可以阻止这种LDL介导的KLF2下调。LDL还可以促进转录抑制因子MeCP2与EZH2、KLF2启动子的结合,同时抑制激活因子MEF2的结合导致KLF2启动子区结合的转录调控因子类型的转变来进一步下调内皮KLF2的表达[29]。KLF2上调TM和eNOS可抑制血管炎症及血栓的形成。LDL抑制内皮KLF2的表达从而调节上述KLF2依赖的基因表达,同时促进PAI⁃1表达。PAI⁃1可促进血栓形成,然而其表达又可被KLF2抑制[29]。综上,LDL胆固醇可以通过表观调控下调KLF2,从而增加动脉粥样硬化性心脏病的发病风险,因此,靶向DNA及组蛋白甲基化的抑制剂或许可以成为一种新的治疗方案,用于高胆固醇相关的血管功能障碍疾病的治疗[29],但是鉴于EZH2抑制剂具有诱发心肌病的潜在风险,其应用需要长期监测及进一步的完善[32]。

图5 EZH1/2在心肌纤维化、心肌肥厚发展中的作用
5 EZH1/2在心肌纤维化、心肌肥厚及心室重构中的作用
Zhu 等[33]发现 miR⁃214⁃3p(microRNA⁃214⁃3p)通过直接靶向EZH1/2来抑制成纤维细胞中心脏基因的表达。 miR⁃214⁃3p可以结合到 EZH1/2增强子3'⁃UTRs上,从转录水平上抑制 EZH1/2的表达,进而增强心肌成纤维细胞中过氧化物酶体增殖物激活受体(PPAR⁃γ)的表达,使Ⅰ型胶原基因 Col1a1、Col3a1表达降低,最终减缓血管紧张素Ⅱ诱导的小鼠心肌组织胶原沉积及心肌纤维化。然而,过表达miR⁃214可以导致心肌肥厚及心脏功能失调,其部分原因可能是EZH2 转录水平降低[34-35]。 由此可见,miR⁃214 及EZH2在机体中发挥作用是剂量依赖的。另有研究[36]发现,血管紧张素II可以抑制长链非编码RNA(lncRNA)TINCR 的表达,通过调控 TINCR/CaMKII通路诱发心肌细胞肥厚。在小鼠心肌细胞中,TINCR直接靶向EZH2,后者直接结合到CaMKII的启动子上并诱导H3K27me3,从而抑制CaMKII的表达,减缓心肌细胞肥大。TINCR敲低后可抑制EZH2在CaMKII启动子区域的结合,从而抑制H3K27me3修饰作用,而过表达 TINCR则正好起到相反的作用[36]。 综上,EZH1/2 作为 miR⁃214 和 TINCR 的直接靶点,有望协同miR⁃214和TINCR成为心肌纤维化、心肌肥大及心脏重构的治疗靶点。
在成鼠心脏中,心肌肥厚相关基因(Anp和β⁃MHC)及促纤维化基因(Tgfβ3)的表达均受 EZH2调控的H3K27me3修饰作用所抑制[14]。心室肥厚与胚胎基因(Anp、Bnp和 β⁃MHC等)的重新激活及成人心脏基因(SERCA2a和 α⁃MHC等)的表达抑制有关。染色质相关的非编码 RNA(ncRNA,比如ANRIL、MHM、H19、AS β⁃MHC 等)通过与 EZH2 相互作用来调控心肌肥厚相关基因表达[37]。上调EZH2及组蛋白去乙酰化酶(HDAC)对特异染色质区域的靶向结合,可以直接抑制包括AS β⁃MHC(心肌肌球蛋白重链)和α⁃MHC在内的心肌肥厚应答基因的表达。当EZH2在心肌肥厚相关基因双向启动子(bdP)上的结合及H3K27m3修饰减少时,将导致这些基因(包括 Anp、Bnp、β⁃MHC 和 Tgfβ3 等)的表达增加。 miR⁃208a、miR⁃208b 和 miR⁃499 等 MyomiRs家族非编码RNA通过促进EZH2对染色质的靶向结合及修饰作用,来沉默肥厚相关基因(Anp、Bnp、Tgfβ3、Spp1)的表达,以此来抑制心肌肥厚。组蛋白去乙酰化酶抑制剂(HDACi,如TSA)可以去除组蛋白上的乙酰基。TSA对成人基因α⁃MHC和AS β⁃MHC表达的去抑制作用(削弱小鼠病理性心肌肥厚)与MHC基因中EZH2的结合及H3K27m3修饰释放相关。在肥厚心肌中,胚胎基因Anp和Bnp的表达上调跟EZH2与这些基因的结合减少有关;而成人基因α⁃MHC 和 AS β⁃MHC的抑制则与bdP处EZH2的结合及H3K27m3修饰增加有关。因此,成人心脏中EZH2对染色质的结合对于维持基因表达的内稳态是必须的。TSA可以抑制心肌肥厚相关基因的表达,从而逆转病理性心肌肥厚并改善心肌功能,防止心力衰竭[38]。
也有研究[39]表明,染色质调控因子 Asxl2对于成年心脏功能及正常心脏基因表达的维持是必须的。Asxl2是PcG的活性增强子[40]。Asxl2在缺血性及特发性扩张型心肌病中表达显著下调。Asxl2抑制β⁃MHC表达,其抑制作用涉及EZH2的组蛋白甲基转移酶活性,Asxl2与EZH2共定位在β⁃MHC的启动子上,直接抑制 β⁃MHC 表达[39]。
Six1作为EZH2的直接靶基因也能够诱导心肌细胞肥大和骨骼肌基因表达。Six1通常只在一个短暂时期表达,然后被EZH2永久沉默,否则将导致心脏病变。研究[41]表明,Six1在心脏前体细胞中发挥功能而在心脏分化中保持沉默状态,EZH2可以通过抑制Six1来稳定心脏相关基因的表达,并阻止心脏病理进程。在心脏前体细胞中敲除EZH2后可以激活Six1依赖的骨骼肌基因表达,从而诱导心肌肥厚,降低Six1的基因水平可以修复这种病理状态[41]。所以胚胎前体细胞的表观调节异常可以成为成年后疾病的诱发因素。因此,EZH2直接调控Six1的转录及表达,在维持心脏形态稳定,尤其是防止心肌肥厚中起到了关键的调控作用。
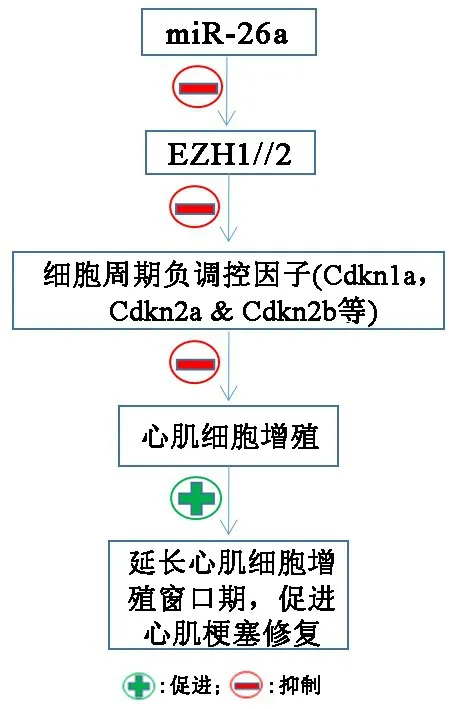
图6 EZH1/2在心肌梗塞修复中的作用
6 EZH1/2在心肌梗塞修复中的作用
miR⁃26a的靶基因(抑制)包括细胞周期激活因子和EZH2,作为miR⁃26a的一个重要靶点,EZH2通过抑制细胞周期的负调控因子的表达来促进心肌细胞增殖,并调控心脏发育中心肌分化相关基因的表达进程[42]。新生小鼠心肌细胞的增殖是以下两方面刺激的结果:miR⁃26a抑制以及EZH2的再表达。在斑马鱼心脏切除后,EZH2被急剧诱导表达,同时伴随miR⁃26a的下调。在小鼠心肌细胞中用经LNA修饰的寡核苷酸抑制miR⁃26a表达后,其靶基因EZH2的表达上调,并显著增强其对发育心脏中的细胞周期负调控因子(包括 Cdkn1a、Cdkn2a和 Cdkn2b)的抑制作用,从而促进心肌细胞的增殖与修复,延长心肌细胞增殖期窗口时间。因此,EZH2/1协同miR⁃26a在修复梗塞心肌中有望成为新的治疗靶点。
7 总结与展望
心脏发育及心血管疾病发生发展受基因水平和表观遗传学水平复杂且精细地调控。本文介绍的EZH1/2作为重要的表观活性因子,通过调控相关转录因子的表达或影响长链非编码RNA(lncRNA)以及microRNA等其他表观遗传机制来调节心脏发育及心血管疾病的进程,是心肌分化、心脏发育及疾病发展中的重要调控因子。作为具有酶活性的功能性蛋白分子,EZH2/1及其修饰产物(H3K27me3等)既可能是心血管发育特定阶段及心血管特定疾病中的关键调控者(原因),也可能是效应因子(结果),无论是“原因”还是“结果”,都可以在疾病治疗、诊断及预防中提供有益的分子理论基础,为心血管疾病的发病机理提供可靠的解决方案。目前,有关EZH1/2与人类心血管发育及疾病的研究还没有系统化,缺乏临床前应用性研究,这可以成为对EZH1/2及其表观调控因子家族进一步研究的方向。
[1]Clowes C,Boylan MG,Ridge LA,et al.The functional diversity of essential genes required for mammalian cardiac development[J].Genesis,2014,52(8):713-737.
[2]Margueron R,Reinberg D.The Polycomb complex PRC2 and its mark in life[J].Nature,2011,469(7330):343-349.
[3]Cardoso C,Timsit S,Villard L,et al.Specific interaction between the XNP /ATR⁃X gene product and the SET domain of the human EZH2 protein[J].Hum Mol Genet,1998,7(4):679-684.
[4]San B,Chrispijn ND,Wittkopp N,et al.Normal formation of a vertebrate body plan and loss of tissue maintenance in the absence of EZH2[J].Sci Rep,2016,6:24658.
[5]Ai S,Yu X,Li Y,et al.Divergent requirements for EZH1 in heart development versus regeneration[J].Circ Res,2017,121(2):106-112.
[6]Kim KH,Roberts CW.Targeting EZH2 in cancer[J].Nat Med,2016,22(2):128-134.
[7]Viré E,Brenner C,Deplus R,et al.The Polycomb group protein EZH2 directly controls DNA methylation [J].Nature,2006,439(7078):871-874.
[8]Buckingham M,Meilhac S,Zaffran S.Building the mammalian heart from two sources of myocardial cells[J].NatRev Genet,2005,6(11):826-835.
[9]龚丁旭,张 浩,胡盛寿.心脏发育过程中常见的相关基因与先天性心脏病[J].基础医学与临床,2011,31(2):207-209.
[10]Coppola A,Romito A,Borel C,et al.Cardiomyogenesis is controlled by the miR⁃99a/let⁃7c cluster and epigenetic modifications[J].StemCell Res,2014,12(2):323-337.
[11]Kataoka M,Huang ZP,Wang DZ.Build a braveheart: the missing linc (RNA)[J].Circ Res,2013,112(12):1532-1534.
[12]Wang QT.Epigenetic regulation of cardiac development and function by polycomb group and trithorax group proteins[J].Dev Dyn,2012,241(6):1021-1033.
[13]Kook H,Seo SB,Jain R.EZ switch from EZH2 to EZH1:Histone methylation opens a window of cardiac regeneration[J].Circ Res,2017,121(2):91-94.
[14]O’Carroll D,Erhardt S,Pagani M,et al.The polycomb⁃group gene Ezh2 is required for early mousedevelopment[J].Mol Cell Biol,2001,21(13):4330-4336.
[15]Chen L,Ma Y,Kim EY,et al.Conditional ablation of Ezh2 in murine hearts reveals its essential roles in endocardial cushion formation,cardiomyocyte proliferation and survival[J].PLoS ONE,2012,7(2):e31005.
[16]He A,Shen X,Ma Q,et al.PRC2 directly methylates GATA4 and represses its transcriptional activity[J].Genes Dev,2012,26(1):37-42.
[17]Pan S,Lai H,Shen Y,et al.DNA methylome analysis reveals distinct epigenetic patterns ofascending aortic dissection and bicuspid aortic valve[J].Cardiovasc Res,2017,113(6):692-704.
[18]Caprio C,Baldini A.p53 Suppression partially rescues the mutant phenotype in mouse models of DiGeorge syndrome[J].Proc Natl Acad Sci USA,2014,111(37):13385-13390.
[19]He A,Ma Q,Cao J,et al.Polycomb repressive complex 2 regulates normal development of the mouse heart[J].Circ Res,2012,110(3):406-415.
[20]Pursani V,Pethe P,Bashir M,et al.Genetic and epigenetic profiling reveals EZH2⁃mediated down regulation of OCT⁃4 involves NR2F2 during cardiac differentiation of human embryonic stem cells[J].Sci Rep,2017,7(1):13051.
[21]Lewandowski SL,Janardhan HP,Trivedi CM.Histone deacetylase 3 coordinates deacetylase⁃independent epigenetic silencing of transforming growth factor⁃β1 (TGF⁃β1) to orchestrate second heart field development[J].J Biol Chem,2015,290(45):27067-27089.
[22]Keck⁃Wherley J,Grover D,Bhattacharyya S,et al.Abnormal microRNA expression in Ts65Dn hippocampus and whole blood:contributions to down syndrome phenotypes[J].Dev Neurosci,2011,33(5):451-467.
[23]Delgado⁃Olguín P,Dang LT,He D,et al.Ezh2⁃mediated repression of a transcriptional pathway upstream of Mmp9 maintains integrity of the developing vasculature[J].Development,2014,141(23):4610-4617.
[24]St⁃Pierre Y,Couillard J,Van Themsche C.Regulation of MMP-9 gene expression for the development of novel molecular targets against cancer and inflammatory diseases[J].Expert Opin Ther Targets,2004,8(5):473-489.
[25]Yang XL,Zhao L,Li SQ,et al.Hyperhomocysteinemia in ApoE-/-mice leads to overexpression of enhancer of zeste homolog 2 via mir⁃92a regulation[J].PLoS ONE,2016,11(12):e0167744.
[26]Lv YC,Tang YY,Zhang P,et al.Histone methyltransferase enhancer of zeste homolog 2⁃mediatedABCA1promoter DNA methylation contributes to the progression of atherosclerosis[J].PLoS ONE,2016,11(6):e0157265.
[27]Greiβel A,Culmes M,Burgkart R,et al.Histone acetylation and methylation significantly change with severity of atherosclerosis in human carotid plaques[J].Cardiovasc Pathol,2016,25(2):79-86.
[28]Wierda RJ,Rietveld IM,van Eggermond MC,et al.Global histone H3 lysine 27 triple methylation levels are reduced in vessels with advanced atherosclerotic plaques[J].Life Sci,2015,129:3-9.
[29]Kumar A,Kumar S,Vikram A,et al.Histone and DNA methylation⁃mediated epigenetic downregulation of endothelial Kruppel⁃like factor 2 by low⁃density lipoprotein cholesterol[J].Arterioscler Thromb Vasc Biol,2013,33(8):1936-1942.
[30]Sako K,Fukuhara S,Minami T,et al.Angiopoietin⁃1 induces Kruppel⁃like factor 2 expression through a phosphoinositide 3⁃kinase/AKT⁃dependent activation of myocyte enhancer factor 2[J].J Biol Chem,2009,284(9):5592-5601.
[31]Huddleson JP,Ahmad N,Lingrel JB.Up⁃regulation of the KLF2 transcription factor by fluid shear stressrequires nucleolin[J].J Biol Chem,2006,281(22):15121-15128.
[32]Katoh M.Mutation spectra of histone methyltransferases with canonical SET domains and EZH2⁃targeted therapy[J].Epigenomics,2016,8(2):285-305.
[33]Zhu WS,Tang CM,Xiao Z,et al.Targeting EZH1 and EZH2 contributes to the suppression of fibrosis⁃associated genes by miR⁃214⁃3p in cardiac myofibroblasts[J].Oncotarget,2016,7(48):78331-78342.
[34]Yang T,Gu H,Chen X,et al.Cardiac hypertrophy and dysfunction induced by overexpression of miR⁃214 in vivo[J].J Surg Res,2014,192(2):317-325.
[35]Yang T,Zhang GF,Chen XF,et al.MicroRNA⁃214 provokes cardiac hypertrophy via repression of EZH2[J].Biochem Biophys Res Commun,2013,436(4):578-584.
[36]Shao M,Chen G,Lv F,et al.LncRNA TINCR attenuates cardiac hypertrophy by epigenetically silencing CaMKII[J].Oncotarget,2017,8(29):47565-47573.
[37]Mathiyalagan P,Keating ST,Du XJ,et al.Interplay of chromatin modifications and non⁃coding RNAs in the heart[J].Epigenetics,2014,9(1):101-112.
[38]Mathiyalagan P,Okabe J,Chang L,et al.The primary microRNA⁃208b interacts with Polycomb⁃group protein,Ezh2,to regulate gene expression in the heart[J].Nucleic Acids Res,2014,42(2):790-803.
[39]Lai HL,Grachoff M,McGinley AL,et al.Maintenance of adult cardiac function requires the chromatin factor Asxl2[J].J Mol Cell Cardiol,2012,53(5):734-741.
[40]Baskind HA,Na L,Ma Q,et al.Functional conservation of Asxl2,a murine homolog for the Drosophila enhancer of trithorax and polycomb group gene Asx[J].PLoS ONE,2009,4(3):e4750.
[41]Delgado⁃Olguín P,Huang Y,Li X,et al.Epigenetic repression of cardiac progenitor gene expression by EZH2 is required for postnatal cardiac homeostasis[J].Nat Genet,2012,44(3):343-347.
[42]Crippa S,Nemir M,Ounzain S,et al.Comparative transcriptome profiling of the injured zebrafish and mouse hearts identifies miRNA⁃dependent repair pathways[J].Cardiovasc Res,2016,110(1):73-84.
