向日葵锈菌转录组SNP位点挖掘及所在基因功能注释
2018-07-06李鑫淳
王 妍,景 岚,李鑫淳
(内蒙古农业大学 农学院,内蒙古 呼和浩特 010019)
锈病是世界范围广泛分布的农作物及森林植物的重大灾害性病害[1-3]。向日葵锈菌(PucciniahelianthiSchw.)属于担子菌门(Basidiomycota)柄锈菌属(Puccinia),它是5种孢子俱全的单主寄生菌。被侵染植株因光合作用受阻以及蒸腾作用加强而引起植株养分和水分供给不足,最终导致向日葵的空壳率增加,果实瘦小,向日葵的含油量和产量下降[4-5]。
目前,对于该病害的防治主要是种植抗病品种,但该病菌可以通过多次无性繁殖产生双核夏孢子进行传播[6],并且可以通过异核作用进行基因重组产生新的致病性小种[7],从而造成病害大流行。近几年来,景岚等[8-9]在向日葵抗锈机制及诱导抗性、形态学解剖、以及转录组学研究等方面展开了工作。随着分子生物学的兴起,已将工作重心转移至对向日葵锈菌系谱、群体进化及遗传多样性研究等方面,如SCAR标记的应用对及微卫星的分析等[10-14]。
单核苷酸多态性(Single nucleotide polymorphisms,SNPs)主要是指基因组核苷酸水平上的变异所引起的DNA序列多态性,同时也指同一物种不同个体间染色体上发生的单碱基的改变[15]。其单个碱基的改变形式主要表现为转换(嘌呤突变为嘌呤或嘧啶突变为嘧啶)、颠换(嘌呤突变为嘧啶或嘧啶突变为嘌呤)以及插入、缺失等。一般情况下,转换比颠换更易发生,转换发生概率为2/3,颠换发生的概率为1/3。对于不同的碱基组成序列SNP发生频率也不同,在GC序列上出现的频率最高,且多发生在C和T之间[16]。SNP是一种常见的可遗传变异,占所有已知多态性的90%以上,已被应用于基因定位、克隆和鉴定。由美国学者 Lander[17]在 1996 年首次提出的SNP 标记是第三代 DNA 遗传标记,相比前几代RFLP和SSR标记,SNP具有突变率低、可稳定遗传的特点,并且其具有双等位基因多态性,使标记更为有效[18-19]。目前,公共可用的 SNP 绝大部分来自大规模基因组测序工作,基于转录组及其他EST序列开发的SNP标记已被广泛应用于玉米、大豆、小麦、人类和寄生虫等的遗传图谱构建和遗传多样性研究[20-24]。
锈菌为专性寄生菌,难以在人工培养基上培养,生活史复杂,具多型现象且遗传转化困难,使得锈菌的基因组学及基因功能研究相对缓慢。近年来,基因组学、转录组学的迅猛发展,为最终揭示锈菌生活史复杂性和毒性高度变异性的根本成因提供了有力工具。
为进一步探究向日葵锈菌的致病机制,本研究利用向日葵锈菌330小种转录组测序得到的数据结合生物信息学软件对锈菌SNP进行较大规模的开发,同时对发现含有SNP位点的基因进行功能注释,认识其基因功能的同时,进一步找到可能与向日葵锈菌致病性相关基因连锁的SNP标记,从而为向日葵锈菌致病性、向日葵锈菌遗传进化研究提供理论基础。
1 材料和方法
1.1 材料及数据来源
由北京博奥公司利用Illumina HiSeqTM2500高通量测序平台对0,4,8 h萌发的向日葵锈菌夏孢子(330小种)进行测序,测序结果用Trinity 软件进行从头组装,共获得386 417 962 个Clean reads,其测序碱基数量为39.03 Gb,拼接获得向日葵锈菌Unigene 59 409个,总长度为82 821 009 bp,其中编码区长37 781 360 bp,非编码区长45 039 649 bp,平均每个Unigene的长度为1 394 bp。测序原始数据已上传至美国国家生物技术信息中心(National Center for Biotechnology Information,NCBI)的SRA数据库( http://www.ncbi.nlm.nih.gov/sra/),数据接收号为SRP 059 519。使用软件SOAPsnp(http://soap.genomics.org.cn/soapsnp.html)对得到的59 409条Unigene序列进行SNP检测。
1.2 试验方法
通过BlastX(E-value<10-5)将含有SNP位点的序列比对到蛋白数据库Nr(Non-redundant protein database)和InterProScan中,然后通过BlastN(E-value<10-5)将此Unigene比对到核酸数据库Nt(Non-redundant nucleotide database)中,得到与给定Unigene具有最高序列相似性的蛋白,从而得到该Unigene的蛋白功能注释信息。
根据Nr注释信息,使用Blast2GO软件得到SNP-unigene的GO(Gene Ontology)注释信息后,用WEGO软件对所有Unigene做GO功能分类统计。通过BlastX(E-value<10-10)将SNP-Unigene序列比对到COG(Clusters of Orthologous Groups of proteins)数据库,从而获得其COG分类注释。通过BlastX(E-value<10-10)将SNP-Unigene序列比对到KEGG生物信息数据库(Kyoto Encyclopedia of Genes and Genomes,http://www.genome.jp/kegg/),根据KEGG注释信息进一步得到Unigene 的pathway注释。最后利用BlastX(E-value<10-10)将SNP-Unigene序列比对到PHI-Base(Pathogen Host Interactions)数据库,从而得到可能与向日葵锈菌致病性相关的蛋白。
2 结果与分析
2.1 向日葵锈菌转录组数据SNP检测
使用SOAPsnp对59 409条Unigene的序列信息进行检测后,共发现SNP位点29 964个,分布在8 321条Unigene上,其中有10 886个在编码区。向日葵锈菌转录组SNP的发生频率为1/2 764 bp,即平均每2 764 bp就有1个SNP位点出现,其中,SNP在编码区的发生频率为1/3 471 bp;在非编码区的发生频率为1/2 361 bp。在总的29 966个SNP中,转换发生的频率为65.40%,颠换发生的频率为34.60%。在这6种单核苷酸变异中以A/G和C/T发生频率最高,分别达到总数的32.80%和32.60%,其他4种A/T、A/C、G/T和G/C则分别占到SNP总数的11.22%,9.18%,8.40%和5.80%(表1)。
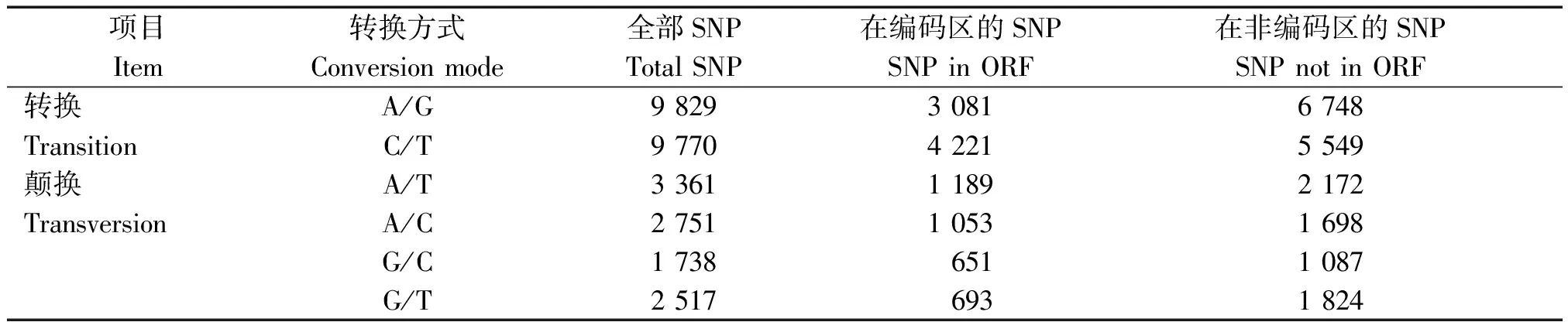
表1 向日葵锈菌SNP 转换、颠换情况统计Tab.1 SNP transition and transversion statistic of P.helianthi
2.2 SNP-Unigene 的功能注释
将所有8 321条SNP-Unigene序列与GenBank中的Nr 数据库进行相似性BlastX比对发现,79.46%的Unigene与数据库中的已知基因同源被注释;通过BlastN与GenBank中的Nt数据库进行相似性比对,结果发现,43.00%的Unigene能在Nt数据库中找到与之匹配的注释信息;与另一蛋白数据库InterProScan进行相似性比对,结果有58.59% 的Unigene被注释,其中包含磷酸酶,各种氧化酶、水解酶等。
2.2.1 SNP-Unigene的GO分类 根据 Nr注释信息,使用Blast2GO软件和 WEGO软件对8 321条SNP-Unigene进行GO功能分类,结果表明,其中3 073条Unigene被分配到38个GO功能分类中,1 134条Unigene被分配到细胞组分的13个子类中,数目最多的类别是参与细胞和细胞部分,占总Unigene数目的比例都为35.70%;2 017条Unigene被分配到分子功能的11个子类中,数目最多的类别是结合和催化,分别为41.23%和34.57%;2 025条Unigene被分配到生物过程中的14个子类中,其中代谢过程和细胞过程所占比例最高,分别为49.04%和45.34%(图1)。

图1 向日葵锈菌SNP-Unigene GO分类Fig.1 GO classification of P.helianthi SNP-Unigene
2.2.2 SNP-Unigene的COG功能分类 COG是对基因产物进行直系同源分类的数据库。将8 321条SNP-Unigene序列比对到COG数据库中进行相关基因功能的预测和分类。结果显示有2 539条Unigene能在COG中找到了相应的注释信息,根据其功能可以被分为24类。从分析统计结果可以看出,这2 539条被注释的Unigene涉及大多数的生命活动过程或功能。一般功能预测类是最大的一个分类,占被注释Unigene的16.27%;其次是翻译、核糖体结构和生物合成与翻译后修饰、蛋白翻转和分子伴侣,分别占比14.45%,12.01%。核结构和细胞迁移2个分类中包含SNP-Unigene最少,分别占比0.08%,0.12%(图2)。
2.2.3 SNP-Unigene的KEGG代谢通路分析 对8 321条Unigene序列进行KEGG代谢通路分析,结果发现,共有2 689条Unigene序列获得了注释,分别参与到物质代谢、遗传信息过程、环境信息过程、细胞学过程、有机体系统、人类疾病六大类生化代谢途径(表2)。分类结果显示,涉及遗传信息过程通路的Unigene最多,占总注释量的42.99%,并以翻译占比最多;其次为物质代谢通路22.16%,并以氨基酸代谢和糖代谢占比最多;17.29%涉及细胞学过程通路,并以转运和分解代谢占比最多;涉及有机体系统通路的Unigene最少,占1.30%。

A.RNA加工和修饰;B.染色质结构和动力学;C.能量的产生和转化;D.细胞周期控制、细胞分裂和染色体分区;E.氨基酸转运和代谢;F.核苷酸转运和代谢;G.碳水化合物转运和代谢;H.辅酶转运和代谢;I.脂类转运和代谢;J.翻译、核糖体结构和生物合成;K.转录;L.复制、重组和修复;M.细胞壁/膜/胞膜生物合成;N.细胞迁移;O.翻译后修饰、蛋白翻转和分子伴侣;P.无机离子转运和代谢;Q.次生代谢物合成、转运和代谢;R.一般功能预测;S.功能未知;T.信号转导机制;U.细胞内运输、分泌和膜泡运输;V.防御系统;Y.核结构;Z.细胞骨架。
A.RNA processing and modification;B.Chromatin structure and dynamics;C.Energy production and conversion;D.Cell cycle control,cell division,chromosome partitioning;E.Amino acid transport and metabolism;F.Nucleotide transport and metabolism;G.Carbohydrate transport and metabolism;H.Coenzyme transport and metabolism;I.Lipid transport and metabolism;J.Translation,ribosomal structure and biogenesis;K.Transcription;L.Replication,recombination and repair;M.Cell wall/membrane/envelope biogenesis;N.Cell motility;O.Posttranslational modification,protein turnover,chaperones;P.Inorganic ion transport and metabolism;Q.Secondary metabolites biosynthesis,transport and catabolism;R.General function prediction only;S.Function unknown;T.Signal transduction mechanisms;U.Intracellular trafficking,secretion,and vesicular transport;V.Defense mechanisms;Y.Nuclear structure;Z.Cytoskeleton.
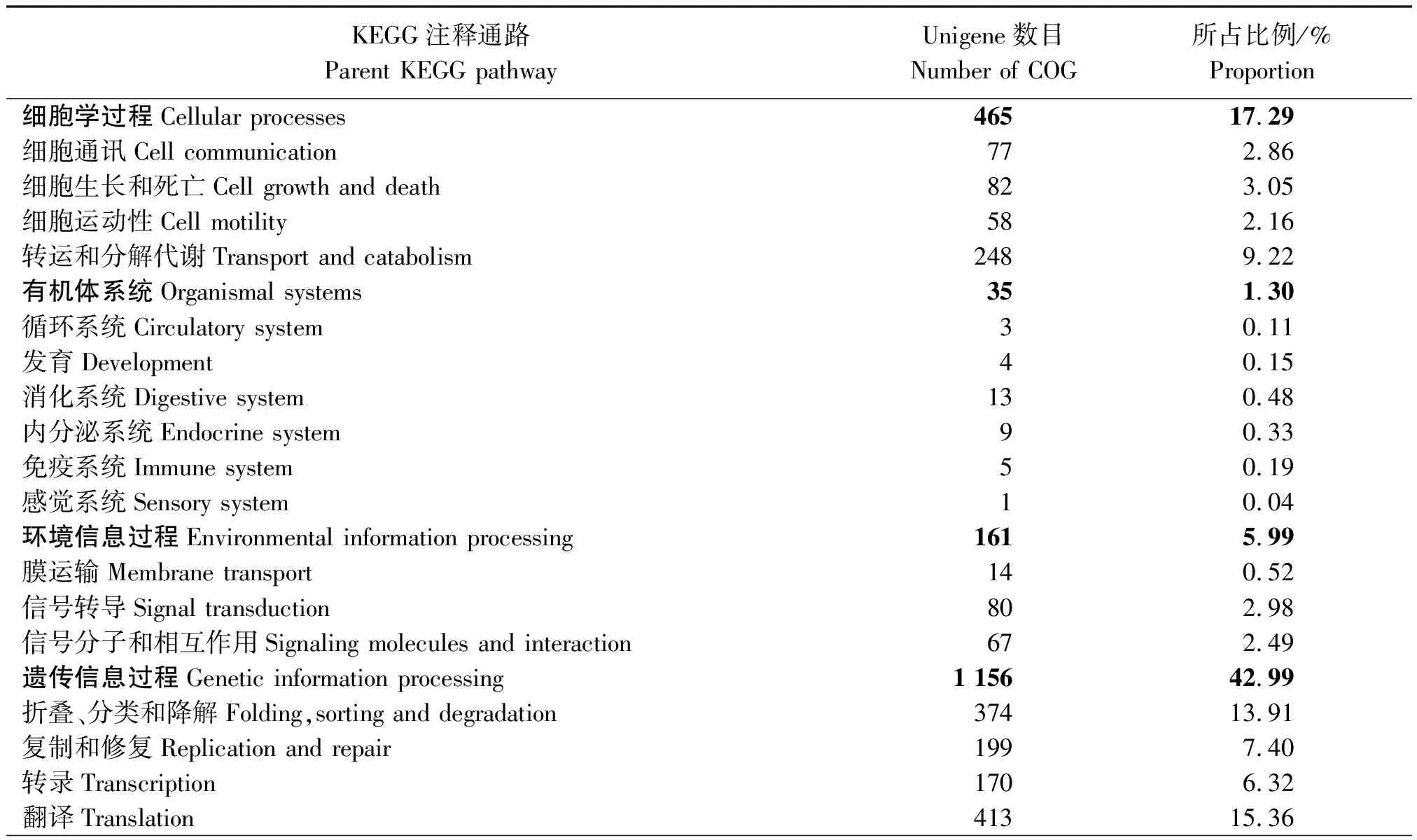
图2 向日葵锈菌SNP-Unigene COG功能分类Fig.2 COG functional classifications of P. helianthi SNP-Unigene

表2(续)
2.2.4 SNP-Unigene的PHI比对 利用BlastX将向日葵锈菌的SNP-Unigene与PHI-Base进行比对,结果显示,有961条Unigene序列与已知的致病基因相匹配(表3),其中,数目最多的为降低致病性基因,占总已知致病基因的44.54%,如热休克蛋白、细胞色素、蛋白磷酸酶2C等;其次是不影响致病力的基因231条,如重复锚蛋白、锌指、核糖体蛋白S17等;丧失致病力基因有118条,如ABC转运蛋白、肽酶C54、蛋白激酶结构域;混合结果80 条;最后是效应因子有4条,将这4条含有效应因子的基因比对到Nr库中,发现只有序列XRKXJ_Cluster 8 762被注释,且与丝氨酸蛋白酶有关。

表3 向日葵锈菌SNP-Unigene PHI分类Tab.3 PHI classifications of P.helianthi SNP-Unigene
3 讨论
基于锈菌基因组巨大、杂合度高[25],向日葵锈菌的SNP开发有其复杂性。与树鼩[26]、苹果[27]、梨[28]、玫瑰[29]、小麦[30]等其他动植物相比,锈菌的遗传背景研究也相对滞后,目前,尚未见有关向日葵锈菌转录组SNP的相关报道。而近年来基于试验数据所建立的生物信息学算法的发展以及向日葵锈菌转录组数据的获得,为转录组水平的向日葵锈菌SNP的开发和应用提供了可能。
本研究对59 409 条向日葵锈菌的Unigene序列进行转录组SNP位点挖掘及其功能注释分析,共鉴定出29 966个SNP,总覆盖长度为 82 821 009 bp,转录组SNP 的发生频率为 1/2 764 bp,高于辣椒(1/6 200 bp)[31],低于人类基因组(约1/1 000 bp),树鼩(1/164 bp)[26]、苹果(1/288 bp)[27]、梨(1/344 bp)[28]、葡萄(1/64 bp)[32]、玫瑰(1/1 173 bp)[29]和小麦(1/540 bp)[30]等大多数动植物。这种频率差异主要与研究材料的遗传背景差异有关,还可能是由检测方法和检测软件参数不同造成的。SNP 频率越高表明遗传背景差异越大[33]。向日葵锈菌SNP在编码区的发生频率为1/3 471 bp,在非编码区的发生频率为1/2 361 bp,由此可见,在编码区发生的频率明显低于非编码区发生的频率,这是在自然选择压力的作用下为保证功能区域在进化过程中的保守性所形成的,符合Syvänen[34]曾经报道的大多数SNPs位于基因组非编码区上,少数位于基因的编码区上的论述。本研究中6种单核苷酸变异以A/G和C/T发生频率最高,分别占总数的32.80%和32.60%,与大多数物种的变异频率基本相似,不仅符合一般的转换、颠换发生频率(即转换发生的概率为2/3,颠换发生的概率是转换的一半为1/3[16]),而且相似于人类基因组的转换、颠换发生频率,而人类基因组中出现这种频率是由于人类基因组中CpG 二核苷酸中的胞嘧啶最容易发生突变,其中大多数是由甲基化自发地脱去氨基而形成胸腺嘧啶[35],因此转换型变异的频率较高,约占2/3,其他几种类型的SNP发生频率基本相同。将8 321条包含SNP位点的Unigene序列比对到蛋白数据库Nr、InterProScan和核酸数据库Nt中,分别有79.46%,58.59%和43.00%的序列被注释。有研究表明,较长的序列越容易得到注释信息,与公共数据越容易匹配[36-37]。本次向日葵锈菌转录组测序最后组装得到的Unigene序列,其平均长度达到1 394 bp,但却不符合这一规律,其中大部分的序列未能被注释,这可能是由于向日葵锈菌是一种专性寄生菌,有其特异性基因,因此与公共数据库较难匹配。
COG注释信息中,向日葵锈菌含有24种COG功能类型,主要包括细胞代谢和细胞信号转导等,其中有2 539个Unigene基因可以匹配到相应的COG功能注释中,经过对各功能类别基因数目进行统计,其中占比较多的类别主要有:一般功能预测;翻译、核糖体结构和生物合成;翻译后修饰、蛋白翻转和分子伴侣等。这些类别对病原菌的调节过程涉及广泛,对细胞的生长、分化、衰老、死亡等都有修饰调节作用,且相互协调、相互影响[38]。这些功能也许与锈菌的生活及侵染有很大的关系,这也符合在KEGG分析中2 689条Unigene注释序列中涉及遗传信息过程通路的Unigene最多,主要以翻译为主。说明蛋白对于病原菌的侵染有十分重要的作用。
真菌遗传图谱的构建工作较高等动植物起步晚,其构建方法主要参考高等动植物。在真菌群中,开发数量充足且多态性好的分子标记用来构建高密度遗传连锁图谱的应用目前还有待深入研究。开发与向日葵锈菌致病性相连锁的SNP分子标记,对向日葵抗病育种的研究具有重大意义。 目前,未见关于向日葵锈菌致病性相关SNP标记的报道。本研究尝试用生物信息学方法,从向日葵锈菌转录组数据中大规模开发分子标记,并筛选可能与致病性相关的SNP标记。Garnica等[39]研究认为,锈菌的致病性是由真菌细胞壁修饰蛋白和潜在的致病效应蛋白所致。病原菌与寄主互作初期通常会产生激发子引发寄主的防卫反应,而大多数激发子属于多糖、多肽、糖蛋白等一类小分子化合物。利用Blast将向日葵锈菌Unigene与PHI-Base中的致病蛋白进行比对,结果发现有961条Unigene与向日葵锈菌的致病性有关,其中有4个基因为植物非病原性基因即效应因子。将这4个效应因子比对到Nr库中,发现序列XRKXJ_Cluster 8 762与丝氨酸蛋白酶有关,而丝氨酸蛋白酶是一类重要的蛋白酶家族,其生物学功能丰富多样,同时还是细菌和真菌病原对抗宿主的重要致病因素[40],并大量产生于病原真菌[41],符合此序列测序时测出的高表达量这一说法。根据测序得到的序列信息进行SNP引物设计,使用CAPS标记法或其他检测方法对开发的SNP进行检测和对向日葵致病性相关SNP的进一步鉴定等工作尚在进行中。向日葵锈菌相关生物信息的不断完善,将为开发SNP遗传多样性引物、遗传图谱的构建、目标基因的标定、指纹图谱绘制等提供理论依据。
参考文献:
[1] Anikster Y,Szabo L J,Eilam T,et al. Morphology,Life cycle biology,and DNA sequence analysis of rust fungi on garlic and chives from California[J]. Phytopathology,2004,94(6):569-577.
[2] 蔡武山,袁 虹,汪友奎,等. 祁连山森林病害防治策略与技术[J]. 林业科技通讯,2015,8:51-54.
[3] 赵 杰,赵世垒,彭岳林,等. 林芝地区小麦条锈菌转主寄主小檗的鉴定与分布[J]. 植物病理学报,2016,46(1):103-111.
[4] 王丽芳. 向日葵抗锈病生理机制及超微结构的研究[D]. 呼和浩特:内蒙古农业大学,2008.
[5] 张文平,张琛平. 巴彦淖尔市向日葵产业发展现状及对策[J].乡村科技,2018(1):10-13.
[6] Middleton K J,Obst N R. Sunflower rust reduces yield[J]. Australian Plant Pathology Society Newsletter,1972,1(3):18-18.
[7] Wright R G. Origin of a new race of puccinia-striiformis[J]. Transactions of the British Mycological Society,1980,74(2):283-287.
[8] 景 岚,王丽芳,康 俊,等. 向日葵品种对锈病抗性的组织学和超微结构研究[J]. 中国油料作物学报,2013,35(3):313-316.
[9] 李凌欣. 寡糖诱导向日葵抗锈病信号转导途径的初步研究[D]. 呼和浩特:内蒙古农业大学,2010.
[10] 胡文婕. 向日葵锈菌夏孢子萌发转录组学研究[D]. 呼和浩特:内蒙古农业大学,2015.
[11] 郭丹丹. 向日葵锈菌分泌蛋白预测及内参基因筛选[D]. 呼和浩特:内蒙古农业大学,2016.
[12] Jing L,Guo D,Hu W,et al. The prediction of a pathogenesis-related secretome ofPucciniahelianthithrough high-throughput transcriptome analysis[J]. BMC Bioinformatics,2017,18(1):166.
[13] 徐 鑫. 向日葵锈病菌的种内群体分化及SCAR标记[D]. 呼和浩特:内蒙古农业大学,2012.
[14] 景 岚,赵诗慧,王 妍,等. 向日葵锈菌夏孢子转录组微卫星特征分析[J]. 中国油料作物学报,2016,38(6):824-830.
[15] Lee Y R,Yoon J B,and Lee J. A SNP-based genetic linkage map ofCapsicumbaccatumandits comparison to theCapsicumannuumreference physical map[J]. Molecular Breeding,2016,36(5):61.
[16] 唐梅森. 基于DNA单核苷酸多态性和甲基化修饰探讨早发冠心病血瘀证的相关易感基因的研究[D]. 长沙:湖南中医药大学,2012.
[17] Lander E S. The new genomics:global views of biology[J]. Science,1996,274(5287):536-539.
[18] 陈 丝,郭 燕,杨 春,等. 单核苷酸多态性在茶树中应用的研究进展[J]. 食品安全质量检测学报,2018,9(2):243-247.
[19] Li R,Li Y,Fang X,et al. SNP detection for massively parallel whole-genome resequencing[J]. Genome Research,2009,19(6):1124-1132.
[20] Frascaroli E,Schrag T A,Melchinger A E. Genetic diversity analysis of elite European maize(ZeamaysL.)inbred lines using AFLP,SSR,and SNP markers reveals ascertainment bias for a subset of SNPs[J]. Theoretical and Applied Genetics,2013,126(1):133-141.
[21] Blair M W,Cortés A J,Penmetsa R V,et al. A high-throughput SNP marker system for parental polymorphism screening,and diversity analysis in common bean(PhaseolusvulgarisL.)[J]. Theoretical and Applied Genetics,2013,126(2):535-548.
[22] 许陶瑜,唐朝晖,王长彪,等. SNP标记在小麦遗传育种中的应用研究进展[J]. 山西农业科学,2017,45(9):1549-1552.
[23] 蒋 刈,戴 朴,韩东一,等. 单核苷酸多态性在人类基因组学发展中的应用[J]. 中华耳科学杂志,2017,15(2):239-244.
[24] 陈 婷,韩红玉,朱顺海,等. 单核苷酸多态性在寄生虫上的应用[J]. 中国动物传染学报,2017,25(6):74-82.
[25] 焦志鑫,申一林,李晶晶,等. 锈菌类真菌基因组结构分析研究进展[J]. 菌物学报,2016,35(12):1456-1467.
[26] 罕园园,孙晓梅,匡德宣,等. 树鼩脂肪转录组SNP位点发掘及其功能注释[J]. 实验动物科学,2016,33(3):13-19.
[27] Chagné D,Crowhurst R N,Troggio M,et al. Genome-wide SNP detection,validation,and development of an 8K SNP array for Apple[J]. PLoS One,2012,7(2):e31745.
[28] 周 贺,李浩男,蔡斌华,等. 砂梨果皮转录组SNP位点发掘及其功能注释分析[J]. 青岛农业大学学报:自然科学版,2014,31(2):105-111.
[29] 梁 芳,张 继,吕 平,等. 基于EST序列的玫瑰EST-SNP位点发掘与分析[J]. 南方农业学报,2016,47(3):325-331.
[30] Somers D J,Kirkpatrick R,Moniwa M,et al. Mining single-nucleotide polymorphisms from hexaploid wheat ESTs[J]. Genome,2003,46(3):431-437.
[31] Lu F H,Yoon M Y,Cho Y I,et al. Transcriptome analysis and SNP/SSR marker information of red pepper variety YCM334 and Taean[J]. Scientia Horticulturae,2011,129(1):38-45.
[32] Lijavetzky D,Cabezas J A,Ibánez A,et al. High throughput SNP discovery and genotyping in grapevine(VitisviniferaL.)by combining a re-sequencing approach and SNPlex technology[J]. BMC Genomics,2007,8:424.
[33] Van Tassell C P,Smith T P,Matukumalli L K,et al. SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries[J]. Nature Methods,2008,5(3):247-252.
[34] Syvänen A C. Accessing genetic variation:genotyping single nucleotide polymorphisms[J]. Nature Reviews Genetics,2001,2(12):930-942.
[35] Collins D W,Jukes T H. Rates of transition and transversion in coding sequences since the human-rodent divergence[J]. Genomics,1994,20(3):386-396.
[36] Shi C Y,Yang H,Wei C L,et al. Deep sequencing of theCamelliasinensistranscriptome revealed candidate genes for major metabolic pathways of tea-specific compounds[J]. BMC Genomics,2011,12:131.
[37] Wang X W,Luan J B,Li J M,et al. De novo characterization of a whitefly transcriptome and analysis of its gene expression during development[J]. BMC Genomics,2010,11:400.
[38] Li S,Zhang L,Yao Q,et al. Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains[J]. Nature,2013,501(7466):242-246.
[39] Garnica D P,Upadhyaya N M,Dodds P N,et al. Strategies for wheat stripe rust pathogenicity identified by transcriptome sequencing[J]. PLoS One,2013,8(6):e67150.
[40] Siezen R J,Leunissen J A. Subtilases:the superfamily of subtilisin-like serine proteases[J]. Protein Science,1997,6(3):501-523.
[41] Dobinson K F,Lecomte N,Lazarovits G. Production of an extracellular trypsin-like protease by the fungal plant pathogenVerticilliumdahliae[J]. Canadian Journal of Microbiology,1997,43(3):227-233.
