Zr、Cr改性CeO2/Al2O3催化剂CO2氧化乙苯脱氢反应研究
2018-07-04冀德坤易玉峰丁福臣刘多强
孙 超,冀德坤,易玉峰,丁福臣,刘多强
(1.北京石油化工学院 燃料清洁化及高效催化减排技术北京市重点实验室,北京 102617;2.中国人民解放军空军油料研究所,北京 100076)
苯乙烯是石油化工行业重要的基础化学品之一。工业生产中,苯乙烯通常是在大量过热水蒸气和高温(高于600 ℃)下由乙苯在铁钾催化剂上脱氢制得[1],但工艺存在能耗过高、水蒸气潜热难以利用和苯乙烯选择性低等缺点。因此,研发新型工艺来解决这些问题十分必要。
乙苯氧化脱氢是近些年的研究热点。大量研究者对O2、SO2、N2、CO2和惰性气体等气氛下的乙苯脱氢反应进行了研究,发现O2气氛下的乙苯氧化脱氢虽然可以降低温度,但易发生乙苯的深入氧化,产生大量的副产物,反应的选择性较低[2-3];以SO2为氧化剂会产生多类副产物,且反应过程中还需考虑防腐、污染等问题;N2和He、Ar等惰性气体气氛下乙苯转化率较低[4-6]。CO2气氛下乙苯脱氢具有加快反应速率、降低能耗、提高苯乙烯选择性等优点,且对CO2的使用消耗有利于降低温室效应等不利影响。因此CO2气氛下的乙苯脱氢工艺是一种绿色环保、节能高效的工艺[7]。
在CO2气氛下的乙苯脱氢反应中,传统催化剂催化效果不好且易失活。CeO2是一种相对便宜且广泛使用的稀土氧化物。Ce具有独特的f电子结构,使CeO2比其他稀土氧化物具有更好的性能[8]。CeO2在催化脱氢方面的优势在于其较强的储放氧能力,而由于纯CeO2高温易烧结、比表面积低等性质[9],CeO2难以直接作为乙苯脱氢催化剂使用,通常加入其他金属氧化物一起使用。
本工作采用沉淀法制备了Zr、Cr改性的CeO2/Al2O3催化剂,利用BET,XRD,SEM,H2-TPR等技术手段对催化剂进行了表征,考察了Zr、Cr改性后催化剂在CO2气氛下的乙苯脱氢性能。
1 实验部分
1.1 催化剂的制备
采用沉淀法制备催化剂。先称取一定量的Ce(NO3)3·6H2O,Zr(NO3)4·5H2O,Cr(NO3)3·9H2O,γ-A12O3于烧杯中,加入适量蒸馏水,搅拌30 min;然后滴如NH3·H2O至pH为10左右,继续搅拌2 h,静置12 h后,真空抽滤得沉淀物,用蒸馏水洗涤沉淀物3次,置于干燥箱中于120 ℃下干燥6 h,最后在550 ℃下焙烧5 h,得催化剂。分别标记为 Ce15Al,Ce15ZrxAl,Ce15CrxAl(x% 为ZrO2或Cr2O3的质量分数)。
1.2 催化剂的表征
BET在北京金埃谱科技有限公司V-Sorb 2800TP型比表面积及孔径分析仪上进行。取150 mg试样在200 ℃下预处理120 min,在77 K液氮温度下通过物理吸附测量催化剂的比表面积。
XRD测试在岛津公司XRD-7000型X射线衍射仪上进行。Cu Kα,管电压40 kV,管电流30 kA,扫描速率 5(o)/min,扫描范围 2θ = 10o~80o。
SEM表征在FEI公司的Quanta 400F型扫描电子显微镜上进行。工作电压30 kV,反应前喷金处理。
H2-TPR表征在浙江泛泰仪器有限公司FINESORB-3010型程序升温化学吸附仪上进行。取10 mg催化剂置于U形石英管中,以10 ℃/min的升温速率升温至400 ℃,在Ar气氛下对催化剂进行30 min的预处理。降温后在5%(φ)H2/Ar气氛下,以10 ℃/min的升温速率升温至800 ℃。
1.3 催化剂的活性评价
催化剂的活性评价在天津大学北洋化工实验设备公司的MR-B-4型催化剂活性评价系统上进行。反应在常压下进行,反应温度600 ℃,CO2与乙苯进料摩尔比10∶1,乙苯进料量用平流泵控制,设为0.1 mL/min,CO2体积流量控制为184 mL/min,催化剂装填量5 g,催化剂尺寸20~40目。
液体产物在福立分析仪器有限公司GC9720型气相色谱仪上进行,Super-QTMPLOT色谱柱,进样口温度200 ℃,柱温160 ℃,检测器温度280 ℃,H2,N2,空气的体积比为1∶1∶10。转化率通过归一化法计算,乙苯转化率(X)和苯乙烯选择性(S)分别由式(1)和(2)计算。
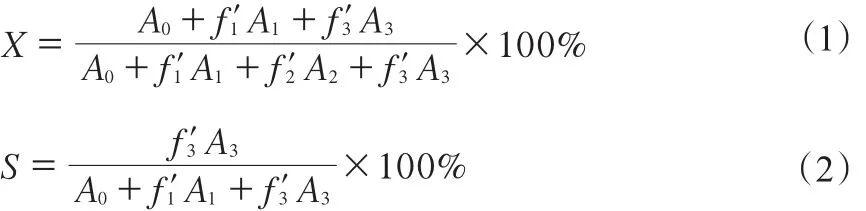
式中,A0,A1,A2,A3分别为苯、甲苯、乙苯和苯乙烯的峰面积;f′1,f′2,f′3分别为甲苯、乙苯和苯乙烯对苯的相对质量校正因子。
2 结果与讨论
2.1 催化剂的活性评价
2.1.1 Ce15ZrxAl催化剂的活性
图1为Zr负载量对CO2气氛下乙苯脱氢反应中乙苯转化率和苯乙烯选择性的影响。由图1可知,当Zr负载量较低时,随着Zr负载量的增加,乙苯转化率提高;当Zr负载量为10%(w)时,乙苯转化率达最高,在反应12 h时乙苯转化率达46%,继续增加Zr负载量,乙苯转化率下降。随Zr负载量的增加,苯乙烯选择性先逐渐增加,在Zr负载量高于12%(w)后选择性变化较小。
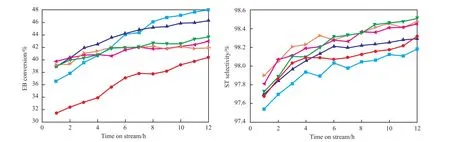
图1 Ce15ZrxAl催化剂的活性Fig.1 Activities of Ce15ZrxAl catalysts.
2.1.2 Ce15CrxAl催化剂的活性
图2为Cr负载量对CO2气氛下的乙苯脱氢反应中乙苯转化率和苯乙烯选择性的影响。由图2可知,当Cr负载量为1%~2%(w)时,催化剂的转化率较低;当负载量大于3%(w)时,催化剂的初始转化率大幅提高,乙苯转化率达55%以上,但转化率下降较快。添加Cr后,苯乙烯的初始选择性降低,但随反应时间的变化幅度较大。综上可知,当Cr负载量为3%(w)时催化剂具有较高的转化率和稳定性,从反应初期乙苯转化率55%到反应12 h后乙苯转化率48.4%。与Ce15A1催化剂相比,Ce15Cr3A1催化剂的乙苯初始转化率较高,而苯乙烯初始选择性较低,稳定性较差。张玉倩[10]对Cr2O3/A12O3催化剂进行了乙苯脱氢实验,也发现Cr2O3/A12O3催化剂初始活性高,但催化剂活性下降速度快。张学彬[11]将Cr助剂添加到V/Al催化剂中进行乙苯脱氢反应也得到相同结论,添加Cr后催化剂的初始活性增加明显,但失活速度明显增加。
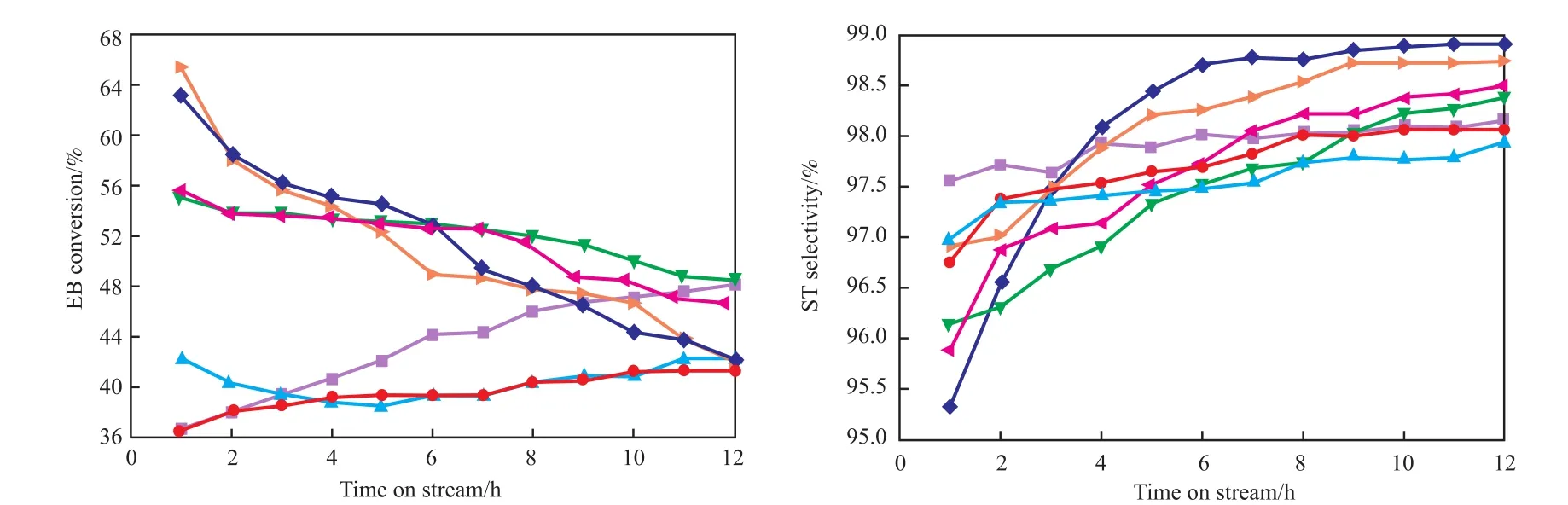
图2 Ce15CrxAl催化剂的活性Fig.2 Activities of Ce15CrxAl catalysts.
为进一步比较Zr和Cr的加入对催化剂催化性能的影响。对Ce15Al,Ce15Zr10Al,Ce15Cr3Al三种催化剂的活性进行考察。图3为Ce15Al,Ce15Zr10Al,Ce15Cr3Al三种催化剂反应36 h内乙苯的转化率和苯乙烯的选择性。由图3可知,Ce15Cr3Al催化剂具有较高的乙苯转化率,在反应前16 h乙苯的转化率较其他2种催化剂有明显的优势,但其乙苯的转化率下降较快,反应到36 h时乙苯转化率仅为32.7%,Ce15Al和Ce15Zr10Al催化剂反应36 h后乙苯转化率分别为35.4%和36.6%,说明Ce15Cr3Al催化剂跟其他两种催化剂相比在长时间反应中不具备优势。Ce15Al和Ce15Zr10Al相比,Ce15Zr10Al催化剂的乙苯初始转化率较好,而Ce15Al催化剂反应初期乙苯的转化率增长速率快,在8 h后高于Ce15Zr10Al催化剂,但当Ce15Al催化剂的乙苯转化率在11 h达到最高后,开始下降,且下降速率较快。Ce15Zr10Al催化剂虽然在反应的中期转化率稍低,但一直较稳定。反应初始阶段,Ce15Cr3Al催化剂的苯乙烯选择性较低,但增长速度快,最终苯乙烯选择性稍高于其他2种催化剂,且3种催化剂的苯乙烯选择性均在98.7%以上。综上可知,Ce15Zr10Al催化剂在长时间反应中具有更好的催化性能。
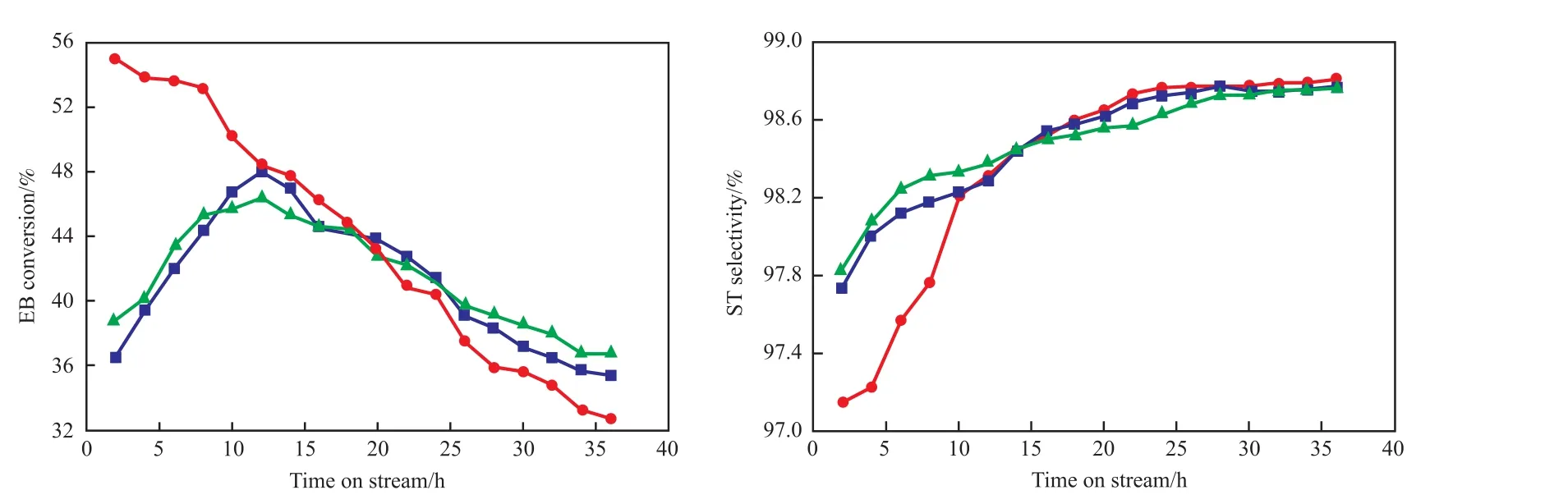
图3 三种催化剂的活性Fig.3 Activities of three catalysts.
2.2 催化剂的表征
2.2.1 BET和积碳分析结果
表 1为 Ce15Al、Ce15Zr10Al和 Ce15Cr3Al催化剂的BET和积碳表征结果。由表1可看出,在Ce15Al催化剂中掺加Cr和Zr后,催化剂的比表面积均有所降低。但Ce15Zr10Al催化剂的比表面积降幅较小,Ce15Cr3Al催化剂相对另两种催化剂的比表面积明显较低。积碳是造成比表面积降低的原因之一,也是导致催化剂失活的重要原因。从反应后积碳量看,反应后3种催化剂均存在明显的积碳,其中,Ce15Zr10Al催化剂在12 h和36 h反应后积碳量均略低Ce15Al催化剂,但总体相差较小;而Ce15Cr3Al催化剂积碳量远高于另两种催化剂,这可能也是Ce15Cr3Al催化剂失活快的重要原因。同时可以发现,反应前12 h积碳较快,随反应时间的延长,积碳减慢。对于Ce15Al和Ce15Zr10Al催化剂,在12 h反应后均有明显的积碳和比表面积降低现象,但在反应12 h内乙苯转化率却处于持续上升的趋势,因此,对这两种催化剂来说,反应初期积碳的存在并没有造成催化剂的失活,其原因可能在于积碳的存在虽然堵塞了催化剂孔道,造成比表面积降低,但同时积碳也具有一定的催化作用,当积碳量较少时反而对催化剂催化性能有一定的促进作用[12]。Nederlof等[13]的研究表明,反应初期产生的积碳有利于提高乙苯转化率和苯乙烯选择性。而当积碳恰好单层覆盖在催化剂表面时催化剂的活性最佳,随后积碳继续沉积,过量的积碳覆盖催化剂表面、堵塞催化剂的孔道,催化剂的活性开始下降。
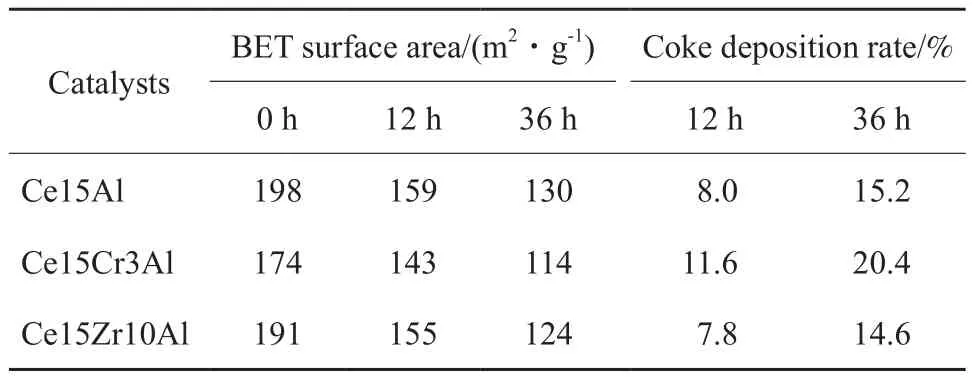
表 1 Ce15Al、Ce15Zr10Al和Ce15Cr3Al催化剂的BET比表面积和积碳率Table 1 BET surface areas and coke deposition amount of Ce15Al,Ce15Zr10Al and Ce15Cr3Al catalysts
2.2.2 XRD分析结果
图 4为 Ce15Al、Ce15Zr10Al和 Ce15Cr3Al催化剂XRD谱图。从图4可看出,对于Ce15Al催化剂,在2θ=36.8o,67.1o处检测到γ-Al2O3的衍射峰,在 2θ=28.6o,33.1o,47.6o,56.3o处检测到 CeO2的衍射峰,分别对应于CeO2的(111)、(200)、(220)、(311)立方萤石结构晶相[14]。没有观察到明显的Ce的其他氧化物的衍射峰,说明催化剂中活性组分为立方萤石结构的CeO2。同时也没有观察到CeO2和A12O3相互作用物质的峰。有研究表明,600 K以下的焙烧温度不利于形成低价Ce的氧化物 CeOx和 CeAlO3[15],673~973 K 的焙烧温度有利于形成非化学计量的CeOx,800 K以上焙烧温度有利于形成CeAlO3。加入Zr和Cr后并没有观察到明显的ZrO2和Cr2O3衍射峰。加入Zr后,催化剂中A12O3的衍射峰强度减小,说明Zr的存在增强了A12O3的分散性,同时衍射峰向高角度移动,这证明了CeO2-ZrO2固溶体的存在,衍射峰角度的移动是因为晶格中Ce4+部分被Zr4+取代,由于Zr4+的离子半径小于Ce4+的离子半径,因此Zr4+进入CeO2晶格中引起晶格收缩,晶格参数变小,使得衍射峰角度向高角度移动[16-17]。同时,催化剂的衍射峰峰形更加尖锐,说明加入Zr后催化剂的晶粒尺寸变大。加入Cr后,催化剂的衍射峰位置没有发生明显变化,催化剂衍射峰峰形变得尖锐,说明催化剂的晶粒变大。叶兴南等[18]对Cr2O3/Al2O3催化剂进行XRD分析,发现当Cr2O3含量高于25%(w)时才会出现明显的Cr2O3的衍射峰。
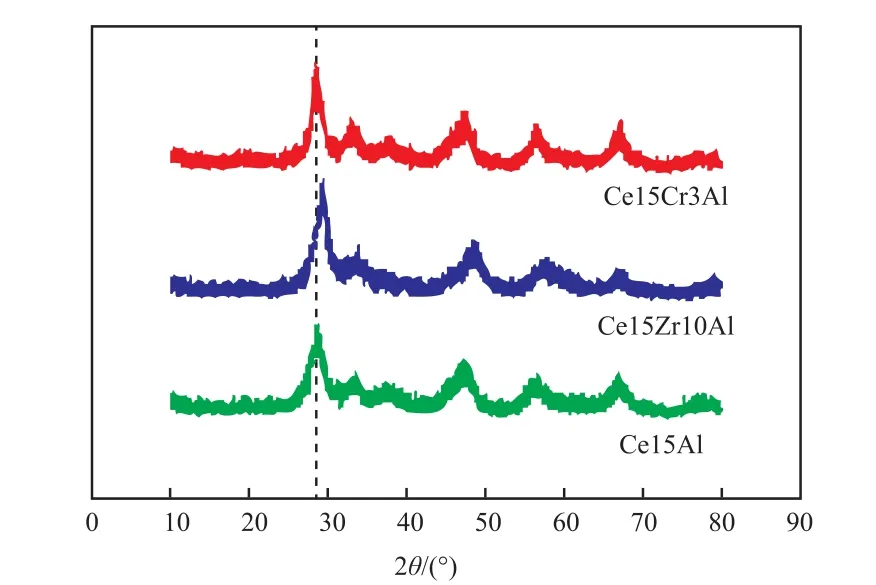
图4 催化剂的XRD谱图Fig.4 XRD patterns of catalysts.
2.2.3 SEM分析结果
图 5为 Ce15Al、Ce15Zr10Al和 Ce15Cr3Al催化剂的SEM照片。由图5a可知,Ce15Al催化剂的颗粒尺寸较小且无固定的几何形态,催化剂存在明显的烧结现象,部分颗粒凝结在一起,分散性较差。由图5b可知,加入Zr后,催化剂的形貌有所变化,催化剂的晶粒尺寸整体变大,这与XRD分析结果相一致,催化剂的颗粒尺寸大小不一,颗粒间分散性较好,催化剂的烧结现象明显改善,说明Zr的加入增加了催化剂的抗烧结能力。而从图5c可知,加入Cr后,催化剂的颗粒粒径明显增大,且存在较严重的团聚现象,颗粒分散不均匀,这可能也是造成Ce15Cr3Al催化剂的比表面积较低的重要原因。

图5 催化剂的SEM照片Fig.5 SEM images of catalysts.
2.2.4 H2-TPR分析结果
图 6为 Ce15Al、Ce15Zr10Al和 Ce15Cr3Al催化剂的H2-TPR谱图。由图6可知,Ce15Al催化剂的H2-TPR谱图中在479 ℃和697 ℃处观察到2个还原峰,分别对应于表面Ce4+的还原和体相Ce4+的还原[19]。在催化剂中加入Zr后没有出现新的衍射峰,这是因为Zr4+难以被还原,同时催化剂表面Ce4+的还原峰强度有所减弱,体相Ce4+的还原峰变得不明显,催化剂的还原性有所降低。这可能也是反应中期Ce15Zr10Al催化活性稍差的原因。Zhang等[17]认为加入Zr后,催化剂的体相晶格氧迁移率降低,这造成了体相Ce4+的还原峰消失,还原性降低,但同时这也是加入Zr后催化剂的热稳定性变好的原因。催化剂中加入Cr后,催化剂的H2-TPR谱图有明显变化,分别在373,477,672 ℃处观察到3个明显的还原峰,分别对应于Cr6+向 Cr3+的还原[20]、表相 Ce4+的还原和体相Ce4+的还原,还原峰面积明显增加,Ce4+还原温度降低,催化剂的还原性增强,这可能也是反应初始阶段Ce15Cr3Al催化剂催化效果好的原因之一。
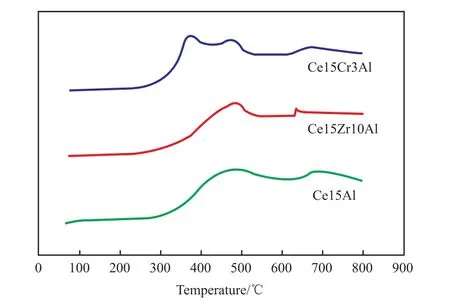
图6 催化剂的H2-TPR谱图Fig.6 H2-TPR spectra of the catalysts.
3 结论
1)Cr改性Ce15Al催化剂,Cr最佳负载量为3%(w),改性后,反应初期乙苯转化率55%,反应12 h后乙苯转化率48.4%;Zr改性Ce15Al催化剂,Zr最佳负载量为10%(w),反应12 h时乙苯转化率达46%。Zr改性的催化剂在长时间的反应中具有更好的催化性能。
2) Cr改性后,Ce15Al催化剂的比表面积明显降低,团聚现象严重;反应后催化剂的积碳率较高,催化剂失活较快。Zr改性后,Zr进入Ce晶格中形成CeO2-ZrO2固溶体,改善了Ce15Al催化剂的烧结现象,催化剂的比表面积和还原性稍有降低,但热稳定性增强,对抑制积碳有积极作用。
[1] 熊丽萍,李国范,谭忠隽,等. 乙苯脱氢制苯乙烯催化剂的研究进展[J].石油化工,2015,44(4):517-522.
[2] 蔡卫权,李会泉,张懿. CO2选择性氧化乙苯制苯乙烯[J].化学进展,2004,16(3):406-413.
[3] Kustrowski P,Segura Y,Chmielarz L,et al. VOxsupported SBA-15 catalysts for the oxidative dehydrogenation of ethylbenzene to styrene in the presence of N2O[J].Catal Today,2006,114(2/3):307-313.
[4] Nederlof C,Talay G,Kapteijn F,et al. The role of RWGS in the dehydrogenation of ethylbenzene to styrene in CO2[J].Appl Catal,A,2012,423/424(8):59-68.
[5] Saito K,Okuda K,Ikenaga N,et al. Role of lattice oxygen of metal oxides in the dehydrogenation of ethylbenzene under a carbon dioxide atmosphere[J].J Phys Chem A,2010,114(11):3845-3854.
[6] Balasamy R J,Tope B B,Khurshid A,et al. Ethylbenzene dehydrogenation over FeOx/(Mg,Zn)(Al)O catalysts derived from hydrotalcites:Role of MgO as basic sites[J].Appl Catal,A,2011,398(1/2):113-122.
[7] 王维维,韩武刚,陈树伟,等. VOx/SBA-16催化CO2氧化乙苯脱氢性能[J].应用化工,2017,46(12):2306-2309.
[8] Xu Jie,Wang Luncun,Liu Yongmei,et al. Mesostructured CeO2as an effective catalyst for styrene synthesis by oxidative dehydrogenation of ethylbenzene[J].Catal Lett,2009,133(3/4):307-313.
[9] 张栖,张明,付名利,等. Al2O3对CexZr1-xO2氧化还原性能的影响[J].中国科技论文在线,2010,5(6):448-452.
[10] 张玉倩. 乙苯CO2脱氢催化剂稳定性的研究[D].上海:华东理工大学,2011.
[11] 张学彬. 钒系催化剂在乙苯混合脱氢过程中的应用[D].西安:陕西师范大学,2008.
[12] Pereira M F R,Orfao J J M,Figueiredo J L. Oxidative dehydrogenation of ethylbenzene on activated carbon fi bers[J].Carbon,2002,40 (13):2393-2401.
[13] Nederlof C,Kapteijn F,Makkee M. Catalysed ethylbenzene dehydrogenation in CO2or N2—Carbon deposits as the active phase[J].Appl Catal,A,2012,417/418(1):163-173.
[14] Rao R,Zhang Qingyun,Liu Huade,et al. Enhanced catalytic performance of CeO2conf i ned inside carbon nanotubes for dehydrogenation of ethylbenzene in the presence of CO2[J].J Mol Catal A:Chem,2012,363/364(11):283-290.
[15] Reddy B M,Rao K N,Reddy G K,et al. Structural characterization and oxidehydrogenation activity of CeO2/Al2O3and V2O5/CeO2/Al2O3catalysts[J].J Phys Chem C,2007,111(50):18751-18758.
[16] 金银龙,王国清,张利军,等. 焙烧温度对铈锆固溶体性能的影响[J].石油化工,2017,46(7):857-861.
[17] Zhang Guizhen,Zhao Zhen,Xu Junfeng,et al. Comparative study on the preparation,characterization and catalytic performances of 3DOM Ce-based materials for the combustion of diesel soot[J].Appl Catal,B,2011,107(3/4):302-315.
[18] 叶兴南,华伟明,乐英红,等. CO2气氛下负载型Cr2O3催化剂上乙苯脱氢制苯乙烯反应[J].催化学报,2004,25(7):581-585.
[19] Li Xiaohong,Feng Jie,Fan Hongxia,et al. The dehydrogenation of ethylbenzene with CO2over CexZr1-xO2solid solutions[J].Catal Commun,2015,59(1):104-107.
[20] 李惠云,乐英红,缪长喜,等. 溶胶-凝胶法制备用于CO2气氛下乙苯脱氢反应的高活性Cr2O3-SiO2催化剂[J].催化学报,2006,27(1):4-6.
