伴眼部异常的TTR型FAP一家系报告及文献复习
2018-07-04胡波琳卢晓庆漆学良丁卫江
胡波琳, 卢晓庆, 漆学良, 丁卫江
家族性淀粉样多神经病变(familial amyloid polyneuropathy,FAP)是一种罕见的常染色体显性遗传病,成人发病,淀粉样物质沉积于全身多个系统,以周围神经系统受累最为明显,诊断的金标准是基因检测。目前研究认为FAP主要是由转甲状腺素蛋白(transthyretin,TTR)、载脂蛋白A-1(apolipoprotein A-1,Apo-A1)和凝溶胶蛋白的变异引起。其中TTR相关FAP(TTR-FAP)最常见,且病情最严重,常危及生命。1984年首次发现该病的致病基因[1]。此后经过研究,目前已发现100多个突变位点[2]。TTR-FAP可累及眼部,主要表现为玻璃体混浊。不过由于对该疾病认识的不足,病理活检的有创性及基因诊断的未广泛普及,目前国内报道的家系较少,推测低于实际发病率,故现将我院收治的1个伴有视物模糊的TTR型FAP家系临床资料和基因检测结果报道如下。
1 资料与方法
患者,男性,33岁,主因“双下肢无力3 m余”就诊于我院。主要表现为走平路和上楼梯时抬腿吃力感,休息数分钟后缓解。夜间偶尔全身肌肉有针刺感和肉跳。既往否认高血压、糖尿病。因视物模糊发现双玻璃体混浊,已行手术治疗。家族中其祖父、父亲、1个叔叔、2个姑姑、2个堂兄弟均有肢体无力及视物模糊,其家系图谱如下(见图1)。患者的堂哥(Ⅲ3)出现四肢无力,双眼玻璃体混浊,对称性非梗阻型肥厚型心肌病;患者的另一个堂哥(Ⅲ1)表现为肢体无力,伴玻璃体出血。入院查体:神志清楚,高级皮质活动正常,双眼球向各方向活动自如,双眼睑闭合有力,双眼视力:右眼0.5,左眼:0.6。双侧面部及肢体痛触觉对称正常,咽反射对称存在,双下肢近端肌力4-级,远端肌力4+级,双上肢肌力5-级。四肢腱反射减低,双下肢痛温觉减退,双下肢肌萎缩明显。病理征阴性,脑膜刺激征阴性。共济运动存在。完善相关检查:血常规:WBC 9.75×109/L(正常值3.5~9.5×109/L),血小板计数124×109/L(正常值125~350×109/L),尿常规、粪便常规、凝血4项、血清葡萄糖、肝肾功能、电解质、乙肝6项、输血4项均未见异常。我院肌电图(EMG)显示广泛性周围神经损害:(1)运动神经远端潜伏期延长、传导速度减慢;双下肢运动神经复合动作电位波幅衰减;感觉神经电位波幅低平,感觉传导速度减慢。(2)正中神经、胫神经刺激所获F波潜伏期延长,波形离散。(3)针级肌电图示较多正相、纤颤电位,轻收缩时双下肢肌肉运动单位电位平均时限增宽,波幅增高。大力收缩时运动单位电位数量减少,呈单纯相。心电图示窦性心率,部分导联ST-T段抬高。胸部平片未见明显异常X线征象。双眼及附属器B超示:双眼玻璃体混浊。双侧眼底照相显示:双眼底出血。双眼OCT示:右眼屈光介质混浊,OCT信号弱,隐约见黄斑区网膜结构大致正常;左眼屈光介质混浊,OCT无信号,未见黄斑区网膜结构。完善右侧腓肠神经病理活检结果示:有髓神经纤维重度缺失伴刚果红阳性淀粉样物质沉积。电镜结果:电镜下胶原纤维增多,有髓神经纤维数目明显减少,部分髓鞘极度松散,神经束膜下见成片聚合物,由长度不等,相互交错排列,淀粉样物质聚合而成,超微结构观察显示:淀粉样变,神经缺失。Ⅲ3、先证者(Ⅲ5)及IV2完善基因检查(送检至北京康旭医学检验所),结果显示:Ⅲ3及先证者TTR基因均出现c.220G>A杂合突变(见图2);Ⅳ2基因结果未见该突变。支持该患者TTR-FAP的诊断。
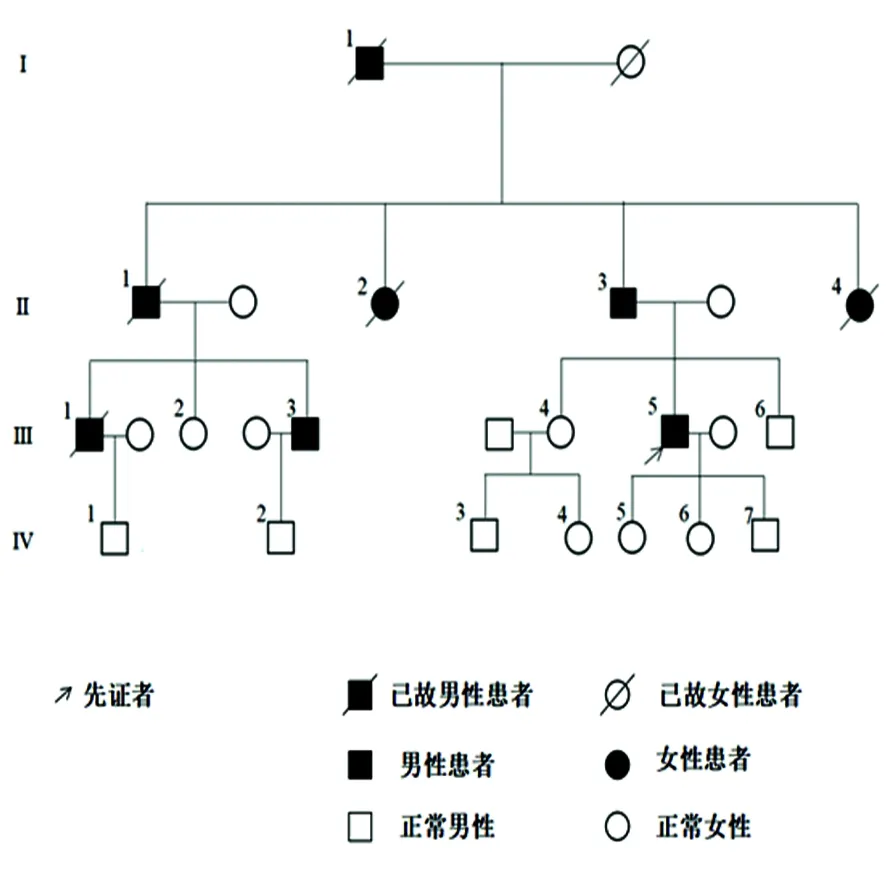
图1 患者家系图谱

图2 先证者(III10)TTR基因检测结果显示18号外显子突变
2 讨 论
TTR-FAP诊断标准:(1)临床诊断:①明确的阳性家族史;②早期发生并且随着病程逐渐进展的多神经病症状;③电生理检查发现大小神经纤维的损害;(2)病病理诊断:刚果红染色发现淀粉样蛋白沉积,免疫组织化学染色显示为TTR淀粉样蛋白;(3)基因诊断:发现特定的TTR基因突变。其中基因诊断为诊断该疾病的金标准[3]。基于上述诊断标准。结合患者的病史、体征、家族史、病理学检查、神经电生理检查及基因检测,该患者有充分的依据诊断为TTR-FAP。
TTR-FAP最常见的眼部病变为玻璃体混浊,混浊物为黄白色棉绒样团块状或片状物质,随时间的延长逐渐增多[4~6]。眼部B型超声检查显示玻璃体团块状回声混浊[7]。眼部异常表现为玻璃体出血的罕见。2007年OHeam TM等报道了2例玻璃体积血患者发生在一个罕见的TTR突变glu54gly中[8],此后TTR-FAP伴玻璃体出血的报道少。在该家系中3人均表现出明显的眼部异常:其中2人伴明显玻璃体混浊;1人伴玻璃体出血。研究发现TTR 可由脉络丛及视网膜色素上皮细胞合成,致病性突变降低了TTR四聚体的稳定性,促进其解离成单体,导致单体在胞外自行聚集,进一步形成寡聚体和淀粉样蛋白,最终发生玻璃体混浊[9]。玻璃体积血形成的原因可能是淀粉样蛋白在视网膜血管壁沉积,阻碍了周围组织的氧输送,缺氧后诱导血管内皮生长因子表达上调;也可能继发于淀粉样浸润所致的血管壁损伤[10]。现于这一家系中发现伴玻璃体出血患者,推测可能是该突变基因所导致的淀粉样蛋白易于沉积于视网膜血管壁,导致最后的出血。该家系中共3个患者出现明显的眼部异常,1个出现玻璃体出血,发生玻璃体出血的概率为(1/3),发生率高于目前的报道。推测可能是因为少量或者微量玻璃体出血被明显的玻璃体混浊掩盖了,导致玻璃体出血目前报道的发生率低于实际发生率。但玻璃体混浊和玻璃体出血可能也是同一种疾病的不同表现形式。随访半年来未再发现玻璃体出血患者,仍需要继续随访。
TTR-FAP是由TTR基因突变导致的常染色体显性遗传病[11],具有基因型和表型异质性[12]。在1984年首次发现该病的突变基因:Val30Met,同时该突变也是目前世纪范围内该病的最常见的突变类型[13]。不过基因的突变仍存在一定的地域差异:在欧洲和日本发病率最高的是Val30Met[14,15]。目前国内报道的与淀粉样变有关的突变有Leu55Arg、Lys35Asn、Ser100Ser、Lys35Thr、Gly83Arg、Arg54Gly等[16]。大多数的TTR-FAP是TTR基因杂合突变所致。在该家系中,出现的也是TTR基因的杂合突变:c.220G>A,即编码区第220号核苷酸由 G 变为 A的杂合核苷酸变异,该变异导致第 74 号氨基酸由 Glu 变为 Lys(p.Glu74Lys)。该突变在人群中发生的频率极低,国外该变异导致的淀粉样多发性神经病已经文献报道[17],而目前中国还未见该突变致病性的报道。这一家系中通过基因检测明确致病的突变为Glu74Lys,虽然目前发现引起的玻璃体淀粉样变性的突变位点众多,但这是目前国内首次发现该突变基因,这一发现增加了我国该疾病的突变基因谱,为将来的该疾病的研究增加了临床数据。并且TTR-FAP患者的外显率并不一定是百分之百,研究发现该疾病在流行区域外显率明显增加:在葡萄牙,Val30Met的外显率达87%,而在瑞典仅为2%[18]。在该家系中有3人完善基因检测,出现基因突变的患者均出现明显的临床症状,外显率为100%。该家系来自中国南方,推测中国南方可能为该疾病的高发区。
TTR-FAP 的治疗相对困难。因为TTR主要在肝脏合成,肝移植是目前最有效的治疗,研究发现肝移植术后患者血清中的变异TTR持续快速下降,病情趋于稳定。至于药物治疗,口服氯苯唑酸可以防止蛋白错误折叠和突变的TTR沉积,已经被批准在欧盟上市[19]。基因疗法及治疗性单克隆抗体尚处于研究阶段但是目前未见肝移植及药物治疗对眼部病变有效性的报道。玻璃体完全切除术是目前治疗该病的有效方法,可显著提高患者的视力,但若切除不完全易导致复发。与此同时,研究发现全视网膜激光光凝可阻止淀粉样物质在玻璃体和视网膜表面沉积。
[参考文献]
[1]Saraiva MJ,Birken S,Costa PP,et al.Amyloid fibril protein in familial amyloidotic polyneuropathy,Portuguese type.Definition of molecular abnormality in transthyretin (prealbumin)[J].J Clin Invest,1984,74(1):104-119.
[2]Benson MD,Kincaid JC.The molecular biology and clinical features of amyloid neuropathy[J].Muscle Nerve,2007,36(4):411-423.
[3]Adams D,Suhr OB,Hund E,et al.First European consensus for diagnosis,management,and treatment of transthyretin familial amyloid polyneuropathy[J].Curr Opin Neurol,2016,29(1):S14-26.
[4]Zou X,Dong F,Zhang S,et al.Transthyretin Ala36Pro mutation in a Chinese pedigree of familial transthyretin amyloidosis with elevated vitreous and serum vascular endothelial growth factor[J].Exp Eye Res,2013,110:44-49.
[5]Liu T,Zhang B,Jin X,et al.Ophthalmic manifestations in a Chinese family with familial amyloid polyneuropathy due to a TTR Gly83Arg mutation[J].Eye(Lond),2014,28(1):26-33.
[6]Kimura A,Ando E,Fukushima M,et al.Secondary glaucoma in patients with familial amyloidotic polyneuropathy[J].Arch Ophthalmol,2003,121(3):351-356.
[7]林海燕,李 莹,杜 虹.家族性淀粉样变多发性神经病变伴双眼多发病变1例[J].中华眼科杂志,2014,50(10):790-791.
[8]OHeam TM,Fawzi A,He S,et al.Early onset vitreous amyloidosis in familial amyloidotic polyneuropathy with a transthyretin Glu54Gly mutation is associated with elevated vitreous VEGF[J].Br J Ophthalmol,2007,91(12):1607-1609.
[9]Merlini G,Bellotti V.Molecular mechanisms of amyloidosis[J].N Engl J Med,2003,349(6):583-596.
[10]Dub EJ,Yang HS,Hailer JA,et al.Vitreous levels of pigment epithelium-derived factor and vascular endothelial growth factor: implications for ocular angiogenesis[J].Am J Ophthalmol,2004,137(4):668-674.
[11]Ando Y,Coelho T,Berk JL,et al.Guideline of transthyretin-related hereditary amyloidosis for clinicians[J].Orphanet J Rare Dis,2013,8:31.
[12]Adams D,Lozeron P,Lacroix C.Amyloid neuropathies[J].Curr Opin Neurol,2012,25(5):564-572.
[13]Plante Bordeneuve V,Said G.Familial amyloid polyneuropathy[J].Lancet Neurol,2011,10(12):1086-1097.
[14]Zaros C,Genin E,Hellman U,et al.On the origin of the transthyretin Val30Met familial amyloid polyneuropathy[J].Ann Hum Genet,2008,72(4):478-484.
[15]Olsson M,Jonasson J,Cederquist K,et al.Frequency of the transthyretin Val30Met mutation in the northern Swedish population[J].Amyloid,2014,21(1):18-20.
[16]王辉林,龙 达,曾 军,等.家族性淀粉样多神经病致病基因突变分析[C].全国医学遗传学学术会议,2009.
[17]谢 兵,蔡善君,蒋 模,等.家族性玻璃体淀粉样变性甲状腺激素结合蛋白Gly83Arg突变家系[J].中华眼底病杂志,2016,32(3):89-91.
[18]Shi Y,Li J,Hu J,et al.A new Arg54Gly transthyretin gene mutation associated with vitreous amyloidosis in Chinese[J].Eye Sci,2011,26(4):230-238.
[19]Misu Ki,Hattori N,Nagamatsu M,et al.Late-onset familial amyloid polyneuropathy type I (transthyretin Met30-associated familial amyloid polyneuropathy) unrelated to endemic focus in Japan.Clinicopathological and genetic features[J].Brain,1999,122(10):1951-1962.
