植物品种SSR指纹分析专用软件SSR Analyser的研发
2018-06-29王凤格李欣杨扬易红梅江彬张宪晨霍永学朱丽葛建镕王蕊任洁王璐田红丽赵久然
王凤格,李欣,杨扬,易红梅,江彬,张宪晨,霍永学,朱丽,葛建镕, 王蕊,任洁,王璐,田红丽,赵久然
植物品种SSR指纹分析专用软件SSR Analyser的研发
王凤格1,李欣2,杨扬1,易红梅1,江彬2,张宪晨2,霍永学2,朱丽2,葛建镕1, 王蕊1,任洁1,王璐1,田红丽1,赵久然1
(1北京市农林科学院玉米研究中心/玉米DNA指纹及分子育种北京市重点实验室,北京 100097,2北京华生恒业科技有限公司,北京 100083)
【目的】开发适用于植物品种SSR指纹分析的软件工具,实现植物品种SSR指纹分析的自动化和标准化,解决SSR标记在实际应用中存在的数据采集效率较低、数据共享难度较大等问题。【方法】在商业化软件GeneMarker®的基础上,针对植物品种SSR指纹分析的特殊性,在SSR指纹处理、panel设计、数据库对接等方面进行算法开发或优化,形成植物品种定制化软件SSR Analyser,并在玉米等多种作物上测试其分析效果。【结果】在SSR指纹处理功能上,软件通过先用系统计算的矩阵进行弱消除,再用Pull-up峰匹配算法进行单峰消除的方案实现了对pull-up峰准确自动消除;通过优化N+1峰、连续多峰等特殊峰型读取的算法,解决了特异峰不识别、读不准、误读等问题,对2 bp重复类型SSR标记为主的植物品种指纹采集更加精准;通过完善邻峰过滤、高低峰过滤和二倍体过滤算法,解决了植物品种混合样品的有效峰采集问题。在Panel设计功能上,软件兼顾了Panel设计的灵活性、方便性和统一性,在保证数据采集标准化的前提下,更加适应复杂的试验情况:提高了标记参数设置的灵活性,根据不同参数作用范围,标记参数设置既有针对特定物种、Panel一次性固定设置的参数,也有针对每个引物位点、每块电泳板单独设定或微调的参数;实现了从已有标记中快速重新组合形成新panel的功能;保证了panel的统一调用和同步更新。通过将软件与指纹库管理系统的无缝对接,实现了样品准备到指纹采集全流程的自动化、标准化,形成的指纹库具有直观可追溯的优势。将SSR Analyser在玉米大规模建库中试用,表明该软件的指纹分析更加简单高效,比原软件分析效率提高了10倍以上;将SSR Analyser扩大到水稻、大豆、黄瓜、西瓜、大白菜等多种二倍体作物和小麦、棉花等多倍体作物中试用,表明在二倍体作物上使用效果较好;在多倍体作物上,对其中的二倍体化的标记使用效果较好,但对非二倍体化的标记仍需进一步完善过滤算法。【结论】开发的SSR Analyser软件具有数据分析程序简单高效、对特殊峰型的SSR标记指纹采集精准、适合基于混合样品的植物品种SSR指纹采集、与指纹库管理系统无缝对接的优点,大大改善了SSR标记在植物品种鉴定中的应用效果。
植物品种;SSR;DNA指纹;荧光毛细管电泳;软件开发
0 引言
【研究意义】SSR标记由于其多态性高、共显性等特点在DNA指纹库构建、品种鉴定及种质资源分析中具有独特的优势[1]。然而,由于SSR标记的等位基因数较多,与只有2个等位基因的SNP、INDEL等标记相比,在实际应用中存在数据采集效率较低、数据共享难度较大等问题[2-3],影响了SSR标记的应用效果。通过采用荧光毛细管电泳平台代替普通凝胶电泳平台,已初步实现了SSR标记在试验程序上的自动化和标准化[4]。如果能进一步通过开发SSR指纹分析工具,实现SSR标记在数据采集上的自动化和标准化,将对推动SSR标记在实践中更加广泛应用具有重要意义。【前人研究进展】目前,SSR指纹分析的常用商业化软件有GeneMapper®和GeneMarker®2个系列[5]。GeneMapper®是ABI公司委托第三方开发的与荧光毛细管电泳仪捆绑销售的DNA片段分析软件,分析对象以人类及马、牛、羊等动物为主[6],针对法庭科学和司法鉴定等应用需求,形成了人类DNA鉴定专用定制版本GeneMapper® ID,主要兼容使用AmpFℓSTR试剂盒[7-8]。GeneMarker®是SoftGenetics公司研发的能够处理主流的荧光毛细管电泳平台输出的DNA数据的商业化软件,面对人类个体鉴定等业务的巨大需求,专为法医学研究提供了人类身份认定STR分型检测软件GeneMarker® HID[9]。可免费获取的分析软件主要有ABI公司提供peak scanner[10],SoftGenetics公司提供的GeneMarker®试用版(http://www.softgenetics. com/GeneMarker.php),以及University of California Davis提供的STRend(http://www.vgl.ucdavis.edu/ informatics/strand.php)等。为解决人类DNA指纹鉴定中的混合样品分析问题,先后开发了MasterMIX[11]、PENDULUM[12]、MIX05[13]、LoComatioN[14]、TrueAllele[15]、Forensim[16]等开放软件以及GeneMapper ®ID- X[17]、GeneMarker®HID[18]等商业化软件。此外,为了商业化客户的需要,除了单机版的分析工具,还出现了一些将分析软件与实验室管理系统等相耦合的操作更加便捷的集成系统[19-20]。【本研究切入点】虽然已经开发了大量的SSR分型软件,特别是GeneMapper®和GeneMarker®系列商业化定制软件在人类DNA指纹库构建及亲子鉴定中得到成功应用,然而,这些软件直接应用到植物品种SSR指纹分析时效果还不太理想,主要原因如下:(1)人类DNA鉴定以单一来源样品的个体识别为主,不同个体的样品混合需作为特殊情况处理[21],植物品种DNA鉴定则以群体识别为主,利用同一品种内部大量不同个体混合形成的混合样品进行DNA指纹分析是主要的样品准备形式,需要将混合样品带来的问题作为普遍问题处理[22];(2)人类的DNA指纹研究比较成熟,SSR标记位点质量高,重复序列以4—5碱基为主,已形成商业化的各种试剂盒产品[23-24],而不同植物物种的DNA指纹研究基础参差不齐,只有少数植物开始参照人类对标记选择的标准开发筛选适合品种鉴定需要的SSR标记[25-26],多数植物入选的SSR标记仍以2碱基重复的标记类型为主,且试验条件标准化程度较低,容易出现连续多峰、N+1峰等特殊峰型,需要更加强大的峰识别算法;(3)人类是二倍体物种,生成的基因分型数据类型较为简单,易于后期的数据分析,而植物物种较多,倍性复杂[27],除了二倍体(如玉米、水稻),还有四倍体(如棉花、油菜)、六倍体(如小麦)等多种类型,增加了指纹采集的难度;(4)人类的DNA指纹库只需要采集数据入库,而植物品种由于其指纹图谱反映的信息量比较大,有同时采集数据和指纹图谱的需求。因此,开发一款适合植物品种SSR指纹分析的软件是非常必要的。【拟解决的关键问题】本研究针对植物品种SSR指纹的特殊性,在GeneMarker的基础上进一步定制适用于植物品种SSR指纹分析的专用软件-SSR Analyser,解决植物品种SSR指纹分析的自动化和标准化的问题,并以玉米等植物的SSR指纹分析为例,验证SSR Analyser的分析性能及应用价值。
1 材料与方法
1.1 供试材料及数据获取
软件开发阶段所用供试材料为玉米品种,参考行业标准《玉米品种鉴定技术规程 SSR标记法》[28]进行DNA提取、PCR扩增、荧光毛细管电泳(DNA分析仪型号:ABI3730XL),通过Data Collection软件将DNA分析仪上获得信号转换为.fsa格式文件,这些原始文件将用于测试软件指纹分析效果。软件试用阶段所用供试材料扩大到水稻[29]、小麦[30]、棉花[31]、高粱[32]、黄瓜[33]、苏丹草[34]等作物品种,所用SSR引物主要从相应作物的行业标准中或试用单位自行筛选的引物名单中选取。
1.2 软件开发整体方案
针对植物品种SSR指纹分析的特殊性,在Genemarker的基础上形成整体定制方案(图1),涉及3个方面:SSR指纹处理、panel编辑、数据库对接。SSR指纹处理是整个软件开发的难点,分为3个环节:原始数据预处理、特异峰识别、有效峰采集。原始数据预处理的研究对象是电泳形成的原始数据,主要包括荧光基线校正、峰图坐标转换、峰的平滑、饱和峰校正、Pull-up峰消除等;特异峰识别的研究对象是原始数据预处理后形成的原始峰,包括单峰、N+1峰、连续多峰等的识别;有效峰采集的研究对象是已识别出的特异峰,包括邻峰过滤、高低峰过滤、二倍体过滤等。Panel编辑主要解决Panel设计、标记参数设置、panel调用等问题。为了实现植物品种指纹库构建及鉴定的全程自动化,将单机版软件进一步与指纹库管理系统进行无缝对接,实现软件直接调用、数据和指纹自动上传、指纹库统一管理和分析等功能。
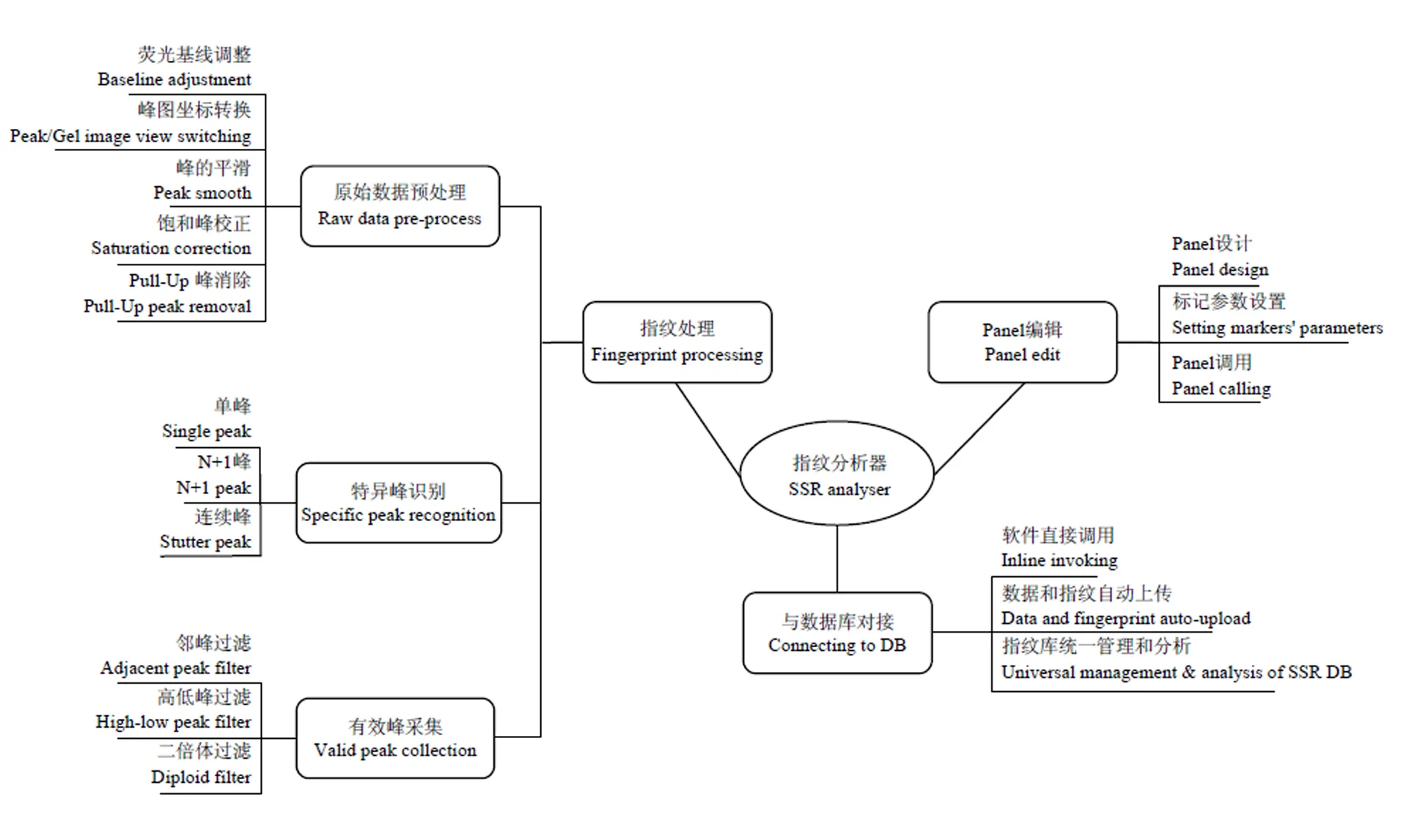
图1 软件开发方案
1.3 SSR指纹处理功能开发
1.3.1 原始数据预处理 原软件对荧光基线校正、峰图坐标转换、饱和峰校正等问题已经解决,因此该环节主要问题是对pull-up峰的识别和消除效果有待改善。Pull-up峰的解决方案有2种:第一种是定义不同荧光之间的作用关系矩阵,按其比例关系对某一颜色的荧光进行整体消除,该方案有2个缺陷,一是不精准,容易产生过度消除或者消除不足;二是荧光之间的作用关系在不同电泳板间是变化的,按固定的比例关系进行消除会出现与实际不符的情况。第二种是结合峰高比例、峰型特征、峰位置等信息,对Pull-up峰进行定位及消除,该方案也有2个缺陷,一是峰高比例偏高时容易过度消除;二是峰型跨度大于分析单元时容易误消。原软件的算法采用了第一种方案,即采用用户自定义的矩阵或系统自动计算的矩阵进行Pull-up峰消除,新软件的算法综合了上述2种方案,即先用系统计算的矩阵进行弱消除,避免过度消除的问题,如果有消除不彻底的情况,再用自定义矩阵进行单峰消除,从而兼顾了自动化和灵活性。
1.3.2 特异峰识别 原始数据经过预处理后,以峰图的形式呈现,需要从中识别出特异峰。特异峰的基本类型有单峰、N+1峰、连续多峰3种。N+1峰、连续多峰是识别的难点,原软件对这两种峰的识别存在位置读不准、峰值估不准、多识别或少识别峰的问题。
N+1峰是 PCR过程中普通Taq酶在非模板扩增片段3'端自动加一个腺苷酸引起的,表现为在同一等位基因位置出现相差1 bp的2个峰[35]。原软件对N+1峰按照读取最高峰的方式进行识别,由于N+1峰按照最高峰出现的位置分为总是左高或总是右高型、时左时右型和接近等高型3种类型,对后2种类型在位置读取时容易造成1 bp的偏差。为此,新软件提供了3种读取方式,即读最高峰、读左峰、读右峰。用户根据标记的N+1峰类型自行设定不同读取方式:对总是左高或总是右高型,设定读最高峰;对接近等高型,固定读左峰或读右峰;对有时左高有时右高型,根据最常出现的情况初始设定读左峰或读右峰,随后针对特定电泳板的实际情况可进行临时调整。无论位置读的是哪个峰,峰值均采集最高峰的。
连续多峰是PCR扩增过程中Taq酶在模板链上滑动引起的,表现为扩增产物长度连续梯度递减的一组峰[36],原软件对连续多峰采取读最高峰的方式进行识别,识别能力较弱,体现在几个方面:(1)连续多峰由于其峰型的特殊性,将一个正常特异峰的高度分散到相差2 bp的多个小峰上,使得单个峰的高度很低,如果采用最高峰的峰值,则该连续多峰容易被随后的高低峰过滤和二倍体过滤算法过滤掉;(2)连续多峰最高峰的位置因样品不同或试验不同而出现变化,造成至少2 bp的读取偏差。新软件形成改进的连续多峰识别算法:将引物扩增区间范围内有3个及以上间隔约2 bp、递增的子峰当成一个整体识别为连续多峰;如果连续多峰由峰高递增的子峰构成,则将其最高峰识别为终点峰,如果连续多峰由峰高接近的子峰构成,以峰高大于最高子峰峰高的特定阈值(默认值是87%)的最右边子峰为其终点峰;将终点峰的位置作为为该连续多峰的读取位置;将连续多峰的所有子峰峰高的累加值作为该连续多峰的峰值并标注在终点峰上,后续的高低峰过滤、二倍体过滤环节均采用此峰值作为过滤依据。
1.3.3 有效峰采集 特异峰识别环节完成后,往往还会有邻峰、高低峰、三峰或多峰的情况,需进一步筛选出可采集入库的有效峰,这就需要一系列过滤掉无效峰的算法,主要包括邻峰过滤、高低峰过滤和二倍体过滤算法。
原软件的过滤算法主要是针对个体样品的指纹采集设计的,在应用到植物品种混合样品的指纹采集时效果不太理想,主要原因在于:个体样品上出现的高低峰主要是PCR扩增时因引物不对称扩增造成的;出现3个或多个峰主要是特异峰识别错误造成的,因此过滤算法主要保证数据采集的准确性。而植物品种混合样品上出现的高低峰、三峰或多个峰,则是样品一致性差和引物不对称扩增两种因素综合作用的结果,过滤算法主要保证数据采集的一致性。为此,新软件对原有过滤算法改进后形成新算法如下:首先执行邻峰过滤,将特异峰中相差2 bp且峰高低于邻峰阈值的低峰过滤掉;然后执行高低峰过滤,将峰高低于高低峰阈值的低峰过滤掉;如果剩下的峰仍有2个以上,进一步执行二倍体过滤算法,将低于二倍体阈值的继续过滤掉,最终剩下1—3个峰。如果是3个峰,对峰高前两位的采集入库,对第三个峰在指纹图谱上标识,但数据不入库,如果是1—2个峰,则直接采集入库。
1.4 Panel编辑
Panel编辑主要包括新panel设计、标记参数设置、panel调用3个方面内容。原软件在panel编辑上存在灵活性、方便性和统一性的欠缺问题:新panel生成时需要先从标记设计开始,不能利用已有panel中的标记进行重组;标记参数设置完成后不能根据具体试验情况进行调整修改;panel的调用比较随意,不同实验员在自己的电脑上分析时,可随时修改panel,造成不同批次分析的数据不具有可比性。
新软件为提高panel设计的方便性,增加了针对已有panel中的标记重组设计的功能,用户进行panel设计时只需遵守标记组合的基本原则(即相同荧光的不同引物范围没有交叉区,不同荧光的引物重叠区最小),就可对已有的标记进行重新组合形成新的panel。
新软件为提高标记参数设置的灵活性,不再对所有参数固定化统一设定,而是区分了不同参数的作用范围,分为3种情况:(1)针对特定物种或panel统一设定的参数,包括读取长度范围、读取峰高范围、高低峰过滤、二倍体过滤;(2)针对每个引物位点单独设定的参数,包括连续多峰过滤、N+1峰过滤、邻峰过滤、Intensity过滤;(3)针对每块电泳板进行微调的参数,包括N+1 峰过滤、Intensity过滤。
新软件为保证不同实验室、不同实验员在同一时期使用完全相同的一套panel,实现建库标准化和数据共享,采取了三节点的panel调用方案,将panel从原来的一种状态或类型区分为3种,即用户自定义的、分析时正在启用的、系统默认的:(1)用户自定义panel与原软件的状态相同,由实验员自行设计、自己使用,可新建、修改或删除,包括从已设定的panel中选择引物自由组合成新的panel。(2)新软件新增了2个状态,一个是系统默认panel,由管理员设计并提供给整个系统同步使用,实验员只能使用,但不能做任何修改和调整,以保证实验员能够采用相同的panel进行分析,并保证panel的实时更新;一个是正在启用panel,实验员可修改其部分参数,但不能自动保存,可进行标记范围的左右整体移动,但不可修改等位基因命名及范围,以兼容不同电泳试验的系统误差。
1.5 与数据库管理系统无缝对接
在软件的功能开发及调试完毕后,将其与北京市农林科学院玉米研究中心开发的植物品种DNA指纹库管理系统(以下简称指纹库系统,软件登记号:2015SR085905)进行无缝对接,主要内容包括:(1)软件直接调用,将软件与指纹库系统链接,在数据库界面下可直接打开软件进行SSR指纹分析,原始文件、panel文件则在指纹库系统中管理和调用。(2)数据和指纹图谱自动上传,软件分析后形成的数据和指纹,直接点击上传,指纹库系统将无效数据和指纹过滤后,对符合条件的自动入库,供下一步分析使用。
2 结果
2.1 软件总体情况
软件基于 Borland公司的Borland C++ Builder 6、在GeneMarker基础定制开发而成,可在Windows(XP及以上)操作系统上运行。计算机硬件环境的基本性能要求为:Pentium®III、1 GHz以上的CPU处理器;512 MB以上的内存;20GB以上的可用硬盘空间。该软件具有界面友好、操作简单、功能强大等优点(图2),已经以SSR指纹分析器(英文名:SSR Analyser)的名称申请并获得了计算机软件著作权(登记号:2015SR161217)。SSR Analyser软件可以从官方网站或QQ群上获取,QQ群名为ssr-analyser,群号为683096128;网站链接为http://ssr-analyser.maizedna.org/或http://ssr-analyser.soft.today,在线文档可以帮助潜在用户学习软件各项功能,安装指南及快速上手指南可以帮助用户安装及使用软件,如果仍有问题,可通过邮箱与作者直接联系。
2.2 SSR指纹处理效果
SSR Analyser软件实现了对pull-up峰准确自动消除;解决了N+1峰、连续多峰等特殊峰型不识别、读不准、误读等问题;通过改进邻峰过滤、高低峰过滤和二倍体过滤算法,解决了植物品种混合样品的有效峰采集问题。图3以玉米SSR引物为例展示了新软件在pullup峰消除(图3-A)、N+1峰识别(图3-B)、连续多峰识别(图3-C)、邻峰过滤(图3-D)、高低峰过滤(图3-E)、二倍体过滤(图3-F)等方面的效果,表明SSR Analyser对植物品种的SSR指纹分析效果有明显改善,满足了基于混合样品的、以2 bp重复类型SSR标记为主的植物品种SSR指纹分析的需求。
2.3 Panel编辑功能改进
SSR Analyser软件较好的兼顾了Panel编辑的灵活性、方便性和统一性,在保证数据采集标准化的前提下,更加适应复杂的试验情况:(1)实现了从已有panel中快速重新组合形成新panel的功能,保证了即使没有使用相同的panel,只要使用了相同的标记就能形成标准化的指纹数据(图4-A);(2)提高了标记参数设置的灵活性,根据不同参数作用范围,标记参数设置既有针对特定物种、panel一次性固定设置的参数,也有针对每个引物位点、每块电泳板单独设定或微调的参数(图4-B);(3)保证了panel的统一调用和同步更新,通过三节点的panel调用方案,将系统统一管理的panel和实验员自己管理的panel分开,和正在运行的可进行参数微调的panel分开,兼顾了panel的稳定性和试验的灵活性(图4-C)。
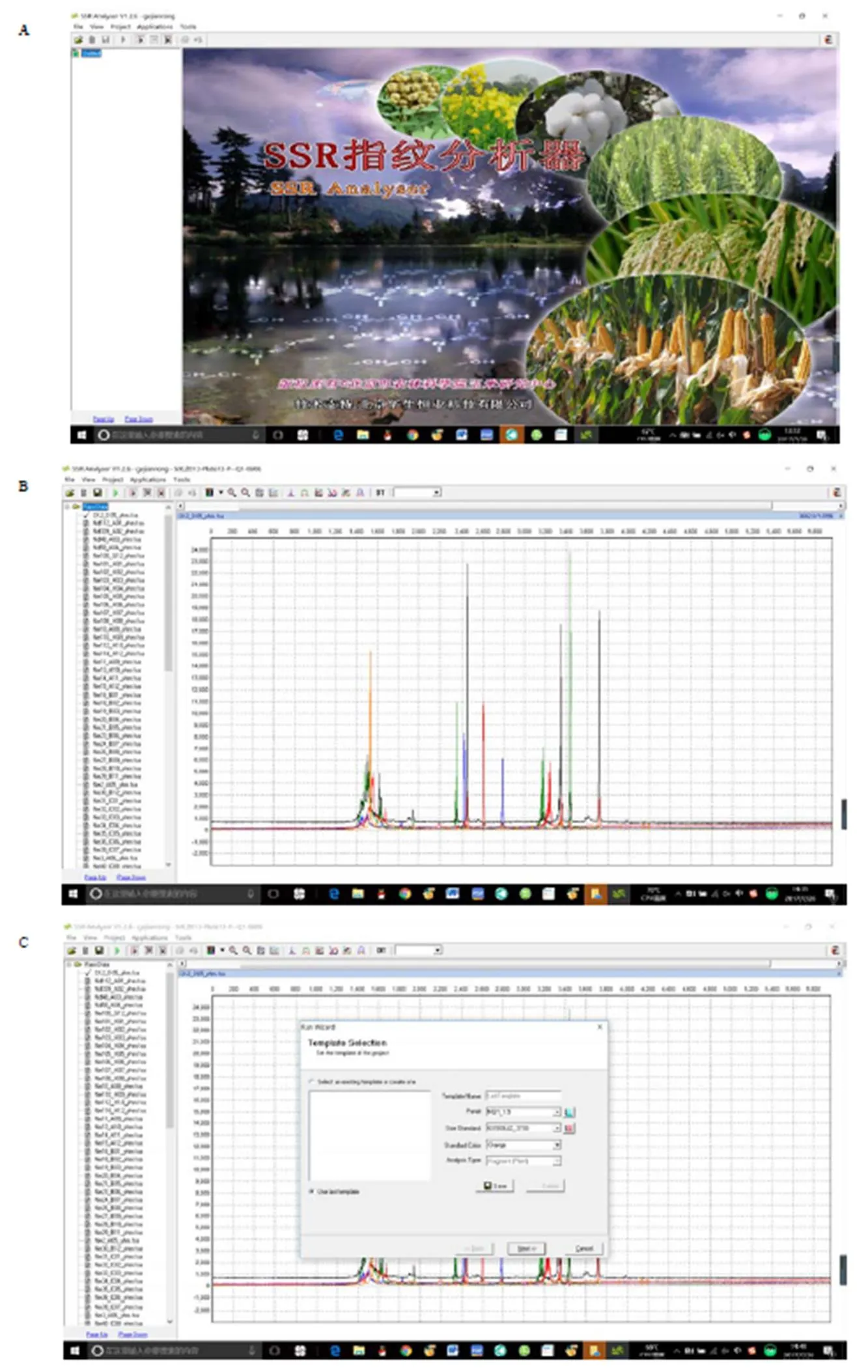
A:主界面;B:原始数据导入;C:参数设置;D:分析工程生成
A: main interface; B: Raw data import; C: Parameter settings; D: creation of analysis project
图2 SSR Analyser软件分析界面
Fig. 2 The user interface of the SSR analyser

A:Pull-up峰消除(以前的算法:pull-up峰由于位于引物扩增区间范围内,未能准确识别并消除;新的算法:pull-up峰识别时考虑到峰型特征,将其准确识别并消除。其中红色底纹表明识别的峰未落入设定的等位基因区间内,灰色底纹表明识别的峰落入设定的等位基因区间内);B:N+1峰识别(以前的算法:由于总是读最高峰,导致位置存在1 bp误差,新的算法:根据该引物N+1峰的特征指定读右峰,避免了位置误差);C:连续多峰识别(以前的算法:该组连续多峰由于峰高过低未能识别;新的算法:成功识别该组连续多峰,峰的高度为所有子峰高度的累加);D:邻峰过滤(以前的算法:未能将相邻的低峰过滤掉;新的算法:将相邻的低于设定阈值的邻峰过滤掉);E:高低峰过滤(以前的算法:未能将低峰过滤掉;新的算法:能够将低峰过滤掉);F:二倍体过滤(以前的算法:仅标识2个峰;新的算法:对符合设定阈值的第三个峰在图上用未加框的灰色底纹标识。)。左侧和右侧的图分别为原软件和新软件的处理效果
A: Pull-up peak elimination (previous algorithm: Because the pull-up peak was located in the range of the primer amplification products, it couldn’t be accurately identified and eliminated; new algorithm: Considering the peak shape feature, it was accurately identified and eliminated. Red shading indicated that the peak did not fall into the allele interval, while grey shading showed that the peak fell into the allele interval); B: N+1 peak recognition (previous algorithm: always read the highest peak, cause 1bp error in position; with new algorithm: read the right peak); C: Tailed peak recognition (pervious algorithm: the height of the tailed peak is too low to be recognized; new algorithm: the tailed peak are successfully identified since the peak height is the summation of all sub peaks height); D: Adjacent peak filtration (Previous algorithms: failed to filter adjacent low peaks; new algorithm: filtered adjacent peaks below the set threshold); E: High-low peak filtration (Previous algorithms: failed to filter low peaks; new algorithm: low peaks couldn’t be filtered out); F: Diploid filtration (Previous algorithms: only two peaks was identified; new algorithm: the third peak that meted the set threshold was marked on the map with no framed grey shade).Graphs on the left show the results of the previous software; graphs on the right show the results of the new software
图3 SSR指纹处理功能的实现效果
Fig. 3 The effect of SSR fingerprint processing function

A:利用已有panel设计新panel;B:标记参数设置;C:Panel使用
A: Designing a new panel with a existing panel; B: Parameter setting of the markers; C: Using the panel
图4 Panel编辑功能的实现效果
Fig. 4 The implementation of the panel editing function
2.4 与指纹库系统对接
SSR Analyser软件与指纹库系统的无缝对接,实现了样品准备到指纹采集全流程的自动化、标准化,形成的指纹库具有直观可追溯的优势(图5)。
2.5 软件实际应用案例
SSR Analyser软件开发后,率先在玉米品种SSR指纹库构建中得到大规模应用[37]。建库SSR引物40个,采用十重电泳,一块电泳板形成的FSA原始文件中包括96个样品、96×10=960个数据点,按10个引物一组设计形成4组系统默认panel(Q1、Q2、Q3和Q4),由两位实验员进行两组独立平行试验并独立进行指纹分析。不考虑数据缺失进行的补板,共形成FSA文件数为42×4×2=336个。首先通过比较两位实验员独立分析的平行试验数据来评估软件分析结果的准确性,两组指纹数据的的吻合度达到99.9%。其次评估软件分析效率,如果用GeneMapper分析一个FSA文件,需要人工订正的数据占10%—20%,平均分析时间为30—40 min,用SSR Analyser分析同样的文件,人工订正的数据量减少为0.5%—1%,平均分析时间缩短为约3 min,两位实验员完成全部336个FSA文件的分析最快需要84 h/人。由此可见,SSR Analyser软件不仅分析准确性高,分析效率也提高了10倍。
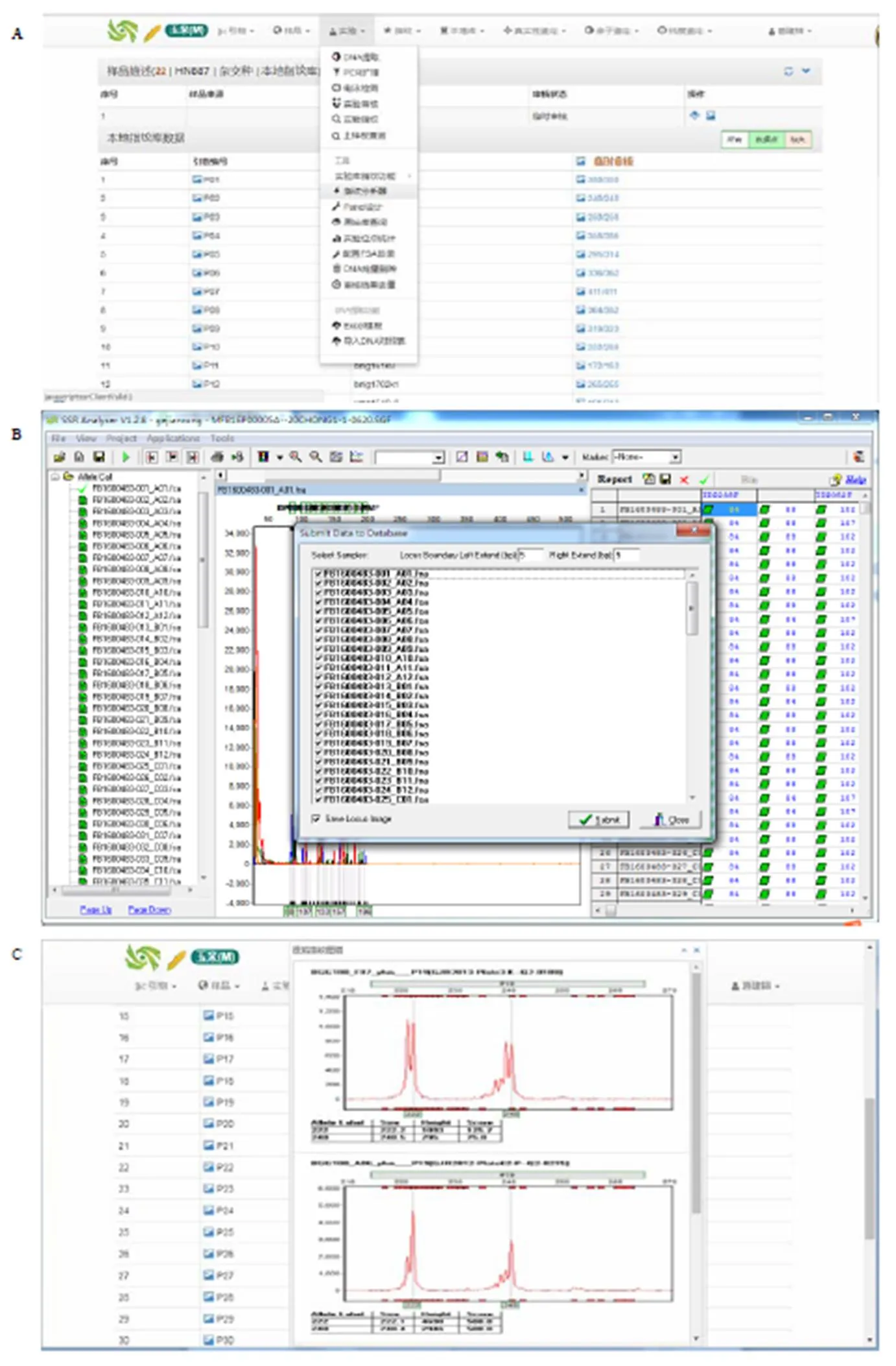
A:在指纹库系统上直接调用分析器;B:分析完毕后直接上传指纹库系统;C:在指纹库系统上管理数据和指纹
为全面系统评价SSR Analyser对不同物种的分析效果,进一步联合多家研究单位在水稻、高粱、黄瓜、番茄、苏丹草等多种二倍体植物的指纹库构建中试用,表明在二倍体作物上的应用是成功的;在小麦、棉花等多倍体植物的指纹库构建中试用,表明对其中二倍体化的标记使用效果较好,对非二倍体化的标记仍需进一步完善过滤算法。
3 讨论
3.1 SSR Analyser软件的应用价值
随着品种审定登记、品种权保护等系列制度的实施,植物品种SSR指纹库构建及品种鉴定已经进入日程。本研究开发的SSR Analyser软件解决了植物品种SSR分析自动化和标准化的问题,与已有的软件相比,更加符合植物品种SSR指纹分析的需求:(1)数据分析更加简单高效,从导入数据、设置参数、分析数据、上传数据,只需连续的四步就可完成,比原软件分析效率提高了10倍以上。(2)对2 bp重复类型SSR标记为主的植物品种指纹采集更加精准,提供了N+1峰、连续多峰等特殊峰型读取的完美解决方案。(3)更加适合基于混合样品的植物品种SSR指纹采集,对混合样品带来的高低峰、三峰、多峰等的指纹采集进行了针对性开发,解决了混合样品的指纹准确读取的问题。(4)与DNA指纹库管理系统自动对接,批量上传数据和指纹图谱。
3.2 软件的物种兼容性
与GeneMapper ID、GeneMarker HID等定制化商业软件仅解决人类一个物种的指纹自动化分析不同,SSR Analyser需要解决大部分植物物种的指纹自动化分析。从SSR Analyser在多个作物上试用的情况看,在二倍体作物上分析效果最好,下一步可在更多的二倍体作物上推广使用。在多倍体作物上的使用效果则受不同SSR标记特征的影响,如果选用的SSR标记类型是二倍体化的,则分析效果较好;如果是多倍体化的,但不同染色体亚组的等位基因落在不同的区间范围内,通过拆解成2个或3个二倍体化的标记,也可获得较好的分析效果。如果无法进行拆解,则面临采集的数据信息不完全的问题。从SSR Analyser软件本身而言,下一步可完善二倍体过滤算法,形成可兼容二倍体、四倍体、六倍体等多种倍性植物的过滤算法;然而,从指纹库构建而言,多倍体过滤算法不仅改变了SSR Analyser的数据采集,还影响到数据库结构设计及品种比较算法的逻辑,因此当开展多倍体植物品种SSR指纹库构建工作时,到底是采取开发二倍体化的SSR标记的方案,还是采取开发多倍体过滤算法的方案,仍需更多的实践检验。
3.3 混合样品的解决方案
人类DNA指纹鉴定多数情况下采集的是个体样品的DNA指纹,个别情况下采集的混合样品主要来自犯罪现场的未知样品,混合样品中混合的不同个体数一般只有2个,即一个未知DNA指纹的个体(嫌疑人)和一个已知DNA指纹的个体(受害者)[11-21],即使如此,对混合样品的指纹解析仍很难达成一致结果[38]。与人类不同,植物品种DNA指纹鉴定可以采集由大量个体混合形成的混合样品,也可以先分别采集多个个体样品,然后统计其主要基因型作为该品种的标准指纹,而混合样品在品种预期一致性较高的情况下是首选方案[22]。与人类上主要处理个体样品或混合2个个体的混合样品的情况不同,SSR Analyser主要处理植物品种混合大量个体的混合DNA,需采取不同的指纹采集算法。在开发策略选择上,采取数据采集和指纹图谱采集相结合的方式,数据采集时通过高低峰过滤和二倍体过滤算法,仅保留峰高排在前面的1—2个峰,对第3个峰在指纹图谱上标注,在上传数据库时,将数据及对应的指纹图谱一并上传,在指纹库管理系统中建立数据和指纹图谱之间的链接,以便于通过指纹图谱获得更详细的信息。从已构建的玉米等作物品种SSR指纹库使用情况看,同时采集数据和指纹大大提升了指纹库的应用价值,在品种真实性鉴定中发挥了重要的作用[37-39]。
4 结论
基于GeneMarker开发了适用于植物品种SSR指纹分析的定制化专用软件-SSR Analyser,解决了植物品种SSR指纹分析的自动化和标准化的问题。与已有软件相比,数据分析程序更加简单高效,对特殊峰型的SSR标记指纹采集更加精准,更加适合基于混合样品的植物品种SSR指纹采集。软件的开发及在多种作物上的成功应用大大改善了SSR标记在植物品种鉴定中的应用效果。
[1] Guichoux E, Lagache L, Wagner S, Chaumeil P, LÉGer P, Lepais O, Lepoittevin C, Malausa T, Revardel E, Salin F, Petit R J. Current trends in microsatellite genotyping., 2011, 11(4): 591-611.
[2] Lü Y, Liu Y, Zhao H. mInDel: a high-throughput and efficient pipeline for genome-wide InDel marker development., 2016, 17(1): 290.
[3] Jones E S, Sullivan H, Bhattramakki D, Smith J S C. A comparison of simple sequence repeat and single nucleotide polymorphism marker technologies for the genotypic analysis of maize (L.)., 2007, 115(3): 361-371.
[4] Sánchez-Pérez R, Ballester J, Dicenta F, Arús P, Martínez-Gómez P. Comparison of SSR polymorphisms using automated capillary sequencers, and polyacrylamide and agarose gel electrophoresis: Implications for the assessment of genetic diversity and relatedness in almond., 2006, 108(3): 310-316.
[5] Phillips N R. Expert systems for high throughput analysis of single source samples: A comparison of GeneMarker® HID v1.71 and GeneMapper® ID v3.2 and Validation of GeneMapper® ID v3.2., 2009.
[6] Chatterji S, Pachter L. Reference based annotation with GeneMapper., 2006, 4(7): R29.
[7] Tsukada K, Harayama Y, Itoga Y, Shimizu M, Kurasawa Y, Kasahara K. Comparison of DNA typing using AmpFlSTR Yfiler and PowerPlex Y System, for specimens subject to very long storage., 2013, 4(1): e162-e163.
[8] Bessetti J. Using GeneMapper® ID with Promega STR Systems., 2005, 8(2): 14-15.
[9] Holland M M, Parson W. GeneMarker® HID: A Reliable software tool for the analysis of forensic STR data., 2011, 56(1): 29-35.
[10] Ream W, Gellar B, Trempy J, Field K. Adding Size Standards to Peak Scanner - Molecular Microbiology Laboratory (Second Edition)-Appendix I. Molecular Microbiology Laboratory: Academic Press, 2013: 197-202.
[11] Gill P, Sparkes R, Pinchin R, Clayton T, Whitaker J, Buckleton J. Interpreting simple STR mixtures using allele peak areas., 1998, 91: 41-53.
[12] Bill M, Gill P, Curran J, Clayton T, Pinchin R, Healy M, Buckleton J. PENDULUM-a guideline-based approach to the interpretation of STR mixtures., 2005, 148(2/3): 181-189.
[13] Slooten K. Validation of DNA-based identification software by computation of pedigree likelihood ratios., 2011, 5(4): 308-315.
[14] Gill P, Kirkham A, Curran J. LoComatioN: A software tool for the analysis of low copy number DNA profiles., 2007, 166(2/3): 128-138.
[15] Perlin M W, Legler M M, Spencer C E, Smith J L, Allan W P, Belrose J L, W D B. Validating TrueAllele® DNA Mixture Interpretation.pdf., 2011, 56(6): 1430-1447.
[16] Haned H. Forensim: an open-source initiative for the evaluation of statistical methods in forensic genetics., 2011, 5: 265-268.
[17] Hansson O, Gill P. Evaluation of GeneMapper® ID-X mixture analysis tool., 2011, 3(1): e11-e12.
[18] He H, Snyder-Leiby T, Qi R, Liu J. Analysis of DNA mixtures in GeneMarker® HID software: with or without single source reference samples., 2009.
[19] Schumm J W, Cunningham H M, Cave C A, Stafford S, Leonard D A. The BodeChecks solution: A high throughput analysis software combining GeneMapper® ID, FSS-i3, LIMS, and artificial intelligence., 2008, 1(1): 125-127.
[20] Rossum T V, Tripp B, Daley D. SLIMS-a user-friendly sample operations and inventory management system for genotyping labs., 2010, 26(14): 1808-1810.
[21] Hu N, Cong B, Li S, Ma C, Fu L, Zhang X. Current developments in forensic interpretation of mixed DNA samples (Review)., 2014, 2(3): 309-316.
[22] 王凤格, 唐浩, 邓超, 周泽宇, 韩瑞玺, 易红梅, 金石桥, 张力科, 赵久然, 吕波, 堵苑苑, 田红丽. NY/T2594-2016 植物品种鉴定DNA分子标记法总则.北京: 中国农业出版社, 2016.
Wang F G, Tang H, Deng C, Zhou Z Y, Hang R X, Yi H M, Jin S Q, Zhang L K, Zhao J R, Lü B, Du Y Y, Tian H L. NY/T2594- 2016. Beijing: China Agriculture Press, 2016. (in Chinese)
[23] White J, Hughes-Stamm S, Gangitano D. Development and validation of a rapid pcr method for the powerplex® 16 hs system for forensic dna identification., 2015, 129(4): 715-723.
[24] Schumm J W, Gutierrez-Mateo C, Tan E, Selden R. A 27-locus STR assay to meet all united states and european law enforcement agency standards., 2013, 58(6): 1584-1592.
[25] 刘文彬, 许理文, 王凤格, 赵久然, 冯博, 赵涵, 吕远大, 蔚荣海. 基于两种荧光毛细管电泳平台筛选评估玉米新型SSR引物. 玉米科学, 2017, 25(2): 24-30.
Liu W B, Xu L W, Wang F G, Zhao J R, Feng B, Zhao H, Lü Y D, Yu R H. Evaluating and screening new maize SSR primer based on two kinds of fluorescent capillary electrophoresis platform., 2017, 25(2): 24-30. (in Chinese)
[26] Bang T C D, Raji A A, Ingelbrecht I L. A multiplex microsatellite marker kit for diversity assessment of large cassava (Crantz) germplasm., 2011, 29(3): 655-662.
[27] Soltis D E, Soltis P S, H L. Molecular data and the dynamic nature of polyploidy., 1993, 12(3): 243-273.
[28] 王凤格, 易红梅, 赵久然, 刘平, 张新明, 田红丽, 堵苑苑. NY/T 1432-2014 玉米品种鉴定技术规程 SSR标记法. 北京: 中国农业出版社, 2014.
Wang F G, Yi H M, Zhao J R, Liu P, Zhang X M, Tian H L, Du Y Y. NY/T 1432-2014. Beijing: China Agriculture Press, 2014. (in Chinese)
[29] 徐群, 魏兴华, 庄杰云, 吕波, 袁筱萍, 刘平, 张新明, 余汉勇, 堵苑苑. NY/T 1433-2014 水稻品种鉴定技术规程 SSR标记法.北京: 中国农业出版社,2014.
Xu Q, Wei X H, Zhuang J Y, Lü B, Yuan Y P, Liu P, Zhang X M, Yu H Y, Du Y Y. NY/T 1433-2014. Beijing: China Agriculture Press, 2014. (in Chinese)
[30] 赵昌平, 支巨振, 邱军, 庞斌双, 刘丽华, 王立新, 谷铁城, 刘丰泽, 吴明生, 刘阳娜, 张立平, 张风廷, 李宏博, 赵海燕. NY/T2859-2015 主要农作物品种真实性SSR分子标记检测普通小麦. 北京: 中国农业出版社, 2015.
Zhao C P, Zhi J Z, Qiu J, Pang B S, Liu L H, Wang L X, Gu T C, Liu F Z, Wu M S, Liu Y N, Zhang L P, Zhang F T, Li H B, Zhao H Y. NY/T2859-2015(L.). Beijing: China Agriculture Press, 2015. (in Chinese)
[31] 杨剑波, 路曦结, 何团结, 陆徐忠, 郑曙峰, 张小娟, 倪金龙. NY/T 2634-2014 棉花品种真实性鉴定 SSR分子标记法.北京: 中国农业出版社, 2014.
Yang J B, Lu X J, He T J, Lu X Z, Zheng S F, Zhang X J, Ni J L. NY/T 2634-2014. Beijing: China Agriculture Press, 2014. (in Chinese)
[32] 李晓辉, 王凤华, 张春宵, 张学军, 周海涛, 郝彩环, 李淑芳, 刘艳芝, 陶蕊, 李万军, 徐宁. NY/T 2467-2013 高粱品种鉴定技术规程 SSR分子标记法.北京: 中国农业出版社,2013.
Li X H, Wang F H, Zhang C X, Zhang X J, Zhou H T, Hao C H, LI S F, Liu Y Z, Tao R, Li W J, Xu N. NY/T 2467-2013. Beijing: China Agriculture Press, 2013. (in Chinese)
[33] 苗晗, 张圣平, 顾兴芳, 王烨, 莫青. NY/T 2474-2013 黄瓜品种鉴定技术规程 SSR分子标记法. 北京: 中国农业出版社, 2013.
Miao H, Zhang S P, Gu X F, Wang Y, Mo Q. NY/T 2474-2013. Beijing: China Agriculture Press, 2013. (in Chinese)
[34] 王杰, 高秋, 杨国锋, 孙娟, 马金星, 冯葆昌. 国审苏丹草和高丹草品种SSR指纹图谱构建及遗传多样性分析. 草地学报, 2016, 24(1): 156-164.
Wang J, Gao Q, Yang G F, Sun J, Ma J X, Feng B C. Fingerprint constructing and genetic diversity analyzing ofand×with SSR markers., 2016, 24(1): 156-164. (in Chinese)
[35] Olejniczak M, Krzyzosiak W J. Genotyping of simple sequence repeats-factors implicated in shadow band generation revisited., 2006, 27(19): 3724-3734.
[36] Guichoux E, Lagache L, Wagner S, Chaumeil P, Léger P, Lepais O, Lepoittevin C, Malausa T, Revardel E, Salin F, Petit RJ. Current trends in microsatellite genotyping., 2011, 11(4): 591-611.
[37] 王凤格, 杨扬, 易红梅, 赵久然, 任洁, 王璐, 葛建镕, 江彬, 张宪晨, 田红丽, 侯振华. 中国玉米审定品种标准SSR指纹库的构建. 中国农业科学, 2017, 50(1): 1-14.
Wang F G, Yang Y, Yi H M, Zhao J R, Ren J, Wang L, Ge J R, Jiang B, Zhang X C, Tian H L, Hou Z H. Construction of an SSR-Based standard fingerprint database for corn variety authorized in China., 2017, 50(1): 1-14. (in Chinese)
[38] Dror I E, Hampikian G. Subjectivity and bias in forensic DNA mixture interpretation., 2011, 51(4): 204-208.
[39] 郑永胜, 张晗, 王东建, 孙加梅, 王雪梅, 段丽丽, 李华, 王玮, 李汝玉. 基于荧光检测技术的小麦品种SSR鉴定体系的建立. 中国农业科学, 2014, (19): 3725-3735.
Zheng Y S, Zhang H, Wang D J, Sun J M, Wang X M, Duan L L, Li H, Wang W, Li R Y. Development of a wheat variety identification system based on fluorescently labeled SSR markers., 2014, 47(19): 3725-3735. (in Chinese)
(责任编辑 李莉)
SSR Analyser:A Special Software Suitable for SSR Fingerprinting of Plant Varieties
WANG FengGe1, LI Xin2, YANG Yang1, YI HongMei1, JIANG Bin2, ZHANG XianChen2, HUO YongXue2, ZHU Li2, GE JianRong1, WANG Rui1, REN Jie1, WANG Lu1, TIAN HongLi1, ZHAO JiuRan1
(1Maize Research Center, Beijing Academy of Agricultural and Forestry Sciences/Beijing Key Laboratory of Maize DNA Fingerprinting and Molecular Breeding, Beijing 100097;2BeijingTodaysoft Limited Company, Beijing 100083)
【Objective】Develop software tools for plant variety identification by SSR fingerprinting to realize the automatic and standardized analysis of plant variety identification, solving problems of low efficiency of data collection and hard for data sharing et al for SSR markers in practice. 【Method】Based on commercialized software GeneMarker®, develop and optimize algorithms to deal with the specialty of SSR fingerprinting analysis for data analysis, panel design, database synchronization et al. SSR Analyser is generated as personalized software and tested on maize and other crops for its effectiveness. 【Result】From the perspective of processing function, the software is able to first weakly eliminate pull-up peak using matrix by system calculation. Then, use matching algorithm with single peak removal method to completely remove pull-up peak automatically and correctly. By optimizing the reading algorithm of N+1 peak, stutter peak et al, unrecognized/inaccurate/mis-reading of special peak is solved. Therefore, it is more accurate for Dinucleotide SSR markers’ fingerprinting. By completing the filtering algorithm of neighboring peak, high and low peak, and diploid crops, it solves the problem of effective peak collection for blended samples. From the perspective of panel design, the software balances the flexibility, convenience and uniformity. On the premise of standardized data collection, the software is more suitable for complicated experiment: enhances the flexibility of marker parameter settings, which include setting parameters at one time for specific species using panels, or setting parameters individually for electrophoresis gel for each primer locus; realizes that generating new panels from existing markers; ensures the unification by calling panels and its synchronous updating. By seamless connecting the software and fingerprinting database management system, it realizes the automation and standardization from sample preparation to fingerprint collection, which makes the database more intuitive and traceable. Using the SSR Analyser for building database of maize indicates that the software is more efficient than the original software, which is 10 times of efficiency; After expanding the SSR Analyser’s application scope to rice, soybean, cucumber, watermelon, Chinese cabbage and other diploid crops, and polyploidy crops such as wheat and cotton, suggests that it is more applicable to diploid crops for polyploidy crops, markers developed based on diploid principle have better application. However, other types of markers need optimization on filtering algorithm. 【Conclusion】SSR Analyser is simple and effective in data analysis, which can accurately collect special peak for SSR markers. It is suitable for fingerprint collection of mixed samples and seamless connects with fingerprint database system, which greatly improves the application of SSR markers in plant variety identification.
plant varieties; SSR; DNA fingerprinting; fluorescent capillary electrophoresis; software development
2018-02-06;
2018-03-28
国家重点研发计划(2017YFD0102001)、北京市科委科技计划课题(Z161100001116089)
王凤格,E-mail:gege0106@163.com。李欣,E-mail:lxwgcool@gmail.com。杨扬,E-mail:caurwx@163.com;王凤格、李欣、杨扬为同等贡献作者。
赵久然,E-mail:maizezhao@126.com
10.3864/j.issn.0578-1752.2018.12.003