基于宏基因组方法分析养猪发酵床微生物组季节性变化
2018-06-28陈倩倩王阶平朱育菁张海峰
陈倩倩,刘 波,王阶平,朱育菁,张海峰
(福建省农业科学院农业生物资源研究所,福州 350003)
微生物发酵床以谷壳、秸秆、锯糠、椰糠等农业副产品作为垫料层,禽畜养殖于其上,日常垫料管理和动物拱翻混合垫料与猪粪尿等排泄物,微生物对混合物进行发酵,进而消纳粪便、去除异味,实现无害化养殖,是一种新型环保养猪技术。微生物发酵床的应用可追溯至明崇祯年间,养殖者以碎草和土为垫圈材料,与粪尿充分混合,形成一种厩肥[1]。1970年,日本建立了以木屑为垫料的微生物发酵床系统[2]。1985年加拿大Biotech公司发明以秸秆为垫料的微生物发酵床系统[3]。20世纪90年代末期,微生物发酵床在日本、中国、韩国及其他东南亚国家推广。此后,研究者一直对发酵床系统进行改良和升级。Chan等[4]为了解决夏季高温的难题,进行了发酵床的改良,提高动物福利。Deininger等[5]采用破碎处理的秸秆作为发酵床垫料,其处理粪便效果和提高动物福利方面优于未处理的秸秆。Kapuinen等[6]测定了不同有机垫料的铺填厚度、承载容量、透气能力、耐压程度等指标,发现将泥炭、木屑和稻秸按一定的比例混合后使用,其效果优于单纯的有机垫料。
微生物发酵床垫料中的微生物消纳处理畜禽粪便,细菌在降解过程中起重要作用。早期发酵床微生物研究主要依赖于传统分离培养法。赵国华等[7]采用常规的分离培养法研究了养猪发酵床垫料中微生物群落构成,芽胞杆菌为垫料中的优势菌,起着降解垫料有机质的作用,随着发酵床寿命的延长,其中的微生物群落多样性逐渐降低。王震等[8]同样发现芽胞杆菌是发酵床垫料中的优势细菌,并参与猪粪降解与发酵床病原生防过程。依靠分离培养技术,大量的除臭菌、猪粪降解菌得以更深入的研究与应用。但该方法操作周期长,过程繁琐,很难完全分离到垫料中的不可培养的厌氧菌及特殊营养类型微生物。脂肪酸标记法与PCR-DGGE技术无分离培养过程,能够较为全面揭示微生物多样性。刘波等[9]采用微生物群落脂肪酸生物标记分析了零排放猪场基质垫层多样性,共检测出37个垫料微生物生物标记。宦海琳等[10]采用PCR-DGGE技术对发酵床垫料基质中的细菌群落多样性进行研究,结果表明垫料中的主要菌群是节杆菌属、放线菌属、芽胞杆菌属和梭菌属等,垫料的组成影响其中的微生物群落结构。随着测序技术的发展,宏基因组测序技术成为研究微生物之间以及微生物与环境因子之间互作的有力工具。该技术绕过传统纯培养技术的瓶颈,研究包含特定生境中微生物的全部遗传信息,更全面地揭示微生物多样性[11]。
发酵床垫料中的微生物菌群可分解猪粪尿、抑制病原菌,是发酵床的核心,其种类和数量对发酵床的运行至关重要。利用宏基因组技术研究发酵床微生物变化,能够揭示微生物群落的细菌种类、数量及结构,对于发酵床降解粪污及除臭等功能研究极其重要。在堆肥研究中,Ren等[12]采用16S rRNA基因序列高通量测序技术分析牛粪堆肥中的微生物组结构与变化,发现主要的细菌为拟杆菌门、变形菌门、厚壁菌门及放线菌门,并且发酵各阶段具有不同的细菌群落。笔者前期研究发现,在发酵的不同阶段,垫料中的优势微生物不同,随着发酵时间延长,棒状杆菌属、芽孢杆菌属、枝芽孢杆菌属、假单胞菌属、放线菌属、乳杆菌属含量增加[13]。但是基于宏基因分析养猪发酵床微生物组季节性变化研究鲜见报道。温度影响细胞内的生化反应,如微生物细胞的酶活性、细胞质膜流动性及环境因子(如物质溶解度)等,进而影响微生物的生长和代谢。因此采用Illumina二代测序技术,研究夏季高温与冬季低温胁迫下发酵床垫料细菌微生物组多样性,可为夏、冬季节发酵床的维护和发酵床猪粪的生物降解提供依据。
1 材料与方法
1.1 材料
实验地点:微生物发酵床位于福建省福清渔溪现代设施农业样本工程示范基地。发酵床厚度为80 cm,微生物组采样季节选择夏季和冬季,发酵床夏季和冬季样本采集方法:夏季样本取自微生物发酵床7月份表层垫料,冬季样本取自同年12月份微生物发酵床表层垫料。采用五点取样法采集,同时进行3次重复采样,混合每次采集的垫料,进行细菌多样性研究。垫料由70%椰糠和30%谷壳粉构成,厚度为80 cm。垫料的管理采用两天旋耕一次,两周翻犁一次,两月深钩一次,并根据垫料湿度的变化调整新鲜垫料的添加。本次夏季和冬季垫料的采样深度为0~15 cm,含水量为45%左右。7月份发酵床室内平均温度为30.13℃,12月份为15.67℃。夏季样本记为s1、s2和s3;冬季样本记为w1、w2和w3。
1.2 方法
1.2.1 微生物发酵床垫料总DNA的提取
按土壤DNA提取试剂盒FastDNA SPIN Kit for Soil的操作指南,称取500 mg垫料样本分别进行总DNA的提取。采用琼脂糖凝胶电泳检测总DNA浓度,稀释至终浓度为1 ng·μL-1开展后续试验。
1.2.2 微生物组16S rDNA V3-V4区测序
采用原核生物16S rDNA基因V3-V4区通用引物 338F(5′-ACT CCT ACG GGA GGC AGC AG-3′)和806R(5′-GGA CTA CHV GGG TWT CTA AT-3′对各垫料样本总DNA进行PCR扩增,PCR反应重复3次。取相同体积混合后进行目的片段回收,所用胶回收试剂盒为AxyPrepDNA凝胶回收试剂盒(Axygen公司)。采用 QuantiFluorTM-ST蓝色荧光定量系统(Promega公司)对回收产物进行定量检测。然后构建插入片段为 350 bp的 Paired-End(PE)文库(TruSeqTMDNA Sample Prep Kit建库试剂盒,Illumina公司),经过Qubit定量和文库检测,HiSeq上机测序(上海美吉)。
1.2.3 微生物组测序数据质控与分析
对测序得到的原始数据进行拼接、过滤,得到有效数据。采用Mothur软件(Version 1.36.1)基于有效数据进行 OTUs(Operational Taxonomic Units)聚类和物种分类分析[14]。采用RDP classifier贝叶斯算法对97%相似水平的OTU代表序列进行分类学分析[15]。从各个OTU中挑选出一条序列作为该OTU的代表序列,将该代表序列与已知物种的16S数据库(Silva,http://www.arb-silva.de)进行物种注释分析;根据每个OTU中序列的条数,得到各个OTU的丰度值[16-17]。反映组间各样品之间的共有及特有OTU数目的Venn图,采用VennDiagram软件生成[18]。
相应的数据释放与SRA(Sequence Read Archive)数据库,序列号分别为 SRR5611215(S1),SRR5611214(S2),SRR5611217(S3),SRR5611216(W1),SRR5611223(W2)和 SRR5611222(W3)。
1.2.4 微生物发酵床细菌群落结构季节变化差异分析
选取含量前9的细菌门和前12的细菌属分别比较两个季节在门和属水平上的差异。采用RDA(冗余分析)法,基于线性模型研究温度、垫料、菌群三者之间的关系。在RDA图内,环境因子用箭头表示,箭头连线和排序轴的夹角代表某个环境因子与排序轴的相关性大小,夹角越小,相关性越高;反之相关性越低。环境因子之间的夹角为锐角时表示两个环境因子之间呈正相关关系,不出现锐角时呈负相关关系。
1.2.5 微生物发酵床细菌群落PICRUSt基因预测
16S功能预测是通过PICRUSt(Phylogenetic Investigation of Communities by Reconstruction of Unobserved States)对OTU丰度表进行标准化[19],根据KEGG数据库的信息,获得各功能基因的丰度,推测微生物群落的功能信息。PICRUSt首先根据微生物16S rRNA信息推测亲缘关系最近的微生物,根据其宏基因组预测其他基因片段的功能,与数据库比对获得微生物群落的代谢功能。经PICRUSt分析,绝大多数微生物预测结构与真实基因功能图谱非常相近,但对于无法用数据库比对获得同源物种信息的微生物则无法预测其功能。分析获得KEGG代谢通路包含3个水平信息,本文采用level2水平进行分析。
2 结果与分析
2.1 微生物发酵床细菌种类季节变化差异
夏季和冬季发酵床6个样本共获得762 923个有效序列。所有样本的稀释曲线接近平台(图1),测序深度已经基本覆盖样本中的所有物种,覆盖率高。韦恩图(图2)显示,夏季样本包含1741种OTUs,冬季样本包含1677种OTUs,夏季样本有更丰富的细菌种类。2组样本共有的OTU有1575个,其中夏季特有OTU 166种,占夏季总OTU的9.5%;冬季特有OTU有102种,占冬季总量的6.1%。夏冬垫料6个样本中共检测到1843种OTU类型,其中夏季样本包含34门,70纲,139目,258科,566属和 889种细菌;冬样本包含28门,59纲,124目,231科,525属和832种细菌。夏季微生物发酵床具有更为丰富和多样的细菌类群。
2.2 微生物发酵床细菌群落结构季节变化差异
微生物发酵床主要的细菌为拟杆菌门、厚壁菌门、变形菌门、放线菌门和糖杆菌门(图3)。夏季样本中,主要细菌为拟杆菌门(28.3%)、厚壁菌门(20.7%)、放线菌门(19.8%)和变形菌门(10.4%)。冬季发酵床垫料的主要细菌为拟杆菌门(31.6%)、厚壁菌门(28.1%)和变形菌门(22.3%)。其中,异常球菌-栖热菌门在微生物发酵床夏季样本中的含量为冬季样本的10倍。夏季垫料中的放线菌门、绿弯菌门和螺旋菌门含量也明显高于冬季样本。而变形菌门在冬季样本中的含量远高于夏季样本,为后者的2倍。拟杆菌门和厚壁菌门含量也明显高于夏季样本。
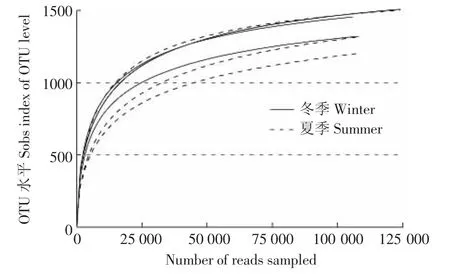
图1 测序样品稀释曲线Figure1 Rarfaction curve of samples
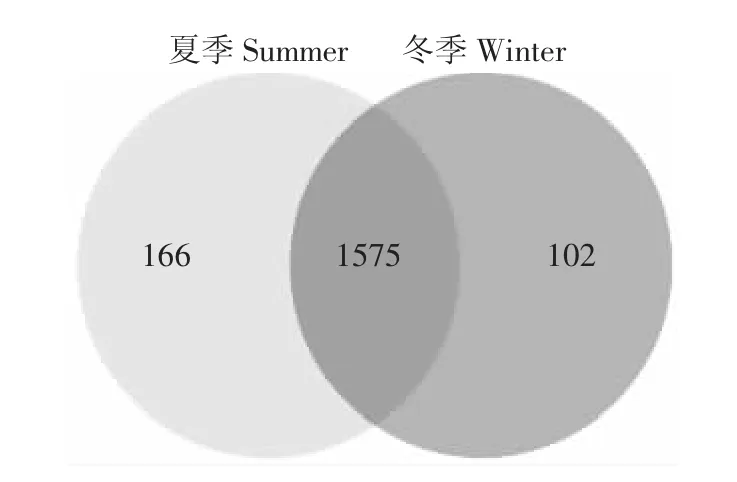
图2 OTU水平的韦恩图Figure2 Venn diagram on OTU level
通过对细菌门水平的研究,发现夏、冬两个季节微生物发酵床垫料中的细菌群落结构不同,细菌分布与温度相适应。夏季中耐热细菌含量多,包括放线菌门、异常球菌-栖热菌门、绿弯菌门和螺旋菌门,并且具有更丰富的细菌种类。冬季,在各种生境中广泛分布的拟杆菌门、厚壁菌门及变形菌门含量高。
在属水平上,两个季节的垫料共检测到581个细菌属,其中夏季样本包含566个属,393 210 reads;冬季样本包含525属,369 713 reads。选取两个季节样本中含量前12的属,比较其在两个季节中的差异(图4)。夏季样本中的主要细菌为糖杆菌属(6.5%)、漠河杆菌属(8.8%)和特吕珀菌属(7.4%)。冬季样本中含量较多的为糖杆菌属(7.1%)、硫假单胞菌(6.8%)、寡源杆菌属(3.5%)和嗜蛋白菌属(3.8%)。异常球菌-栖热菌门的特吕珀菌属在夏季样本中的相对含量为冬季样本的10倍;拟杆菌门的漠河杆菌是冬季样本的13.2倍。假单胞菌科的硫假单胞菌在冬季样本中的相对含量为夏季样本的25倍。发酵床冬季样本中拟杆菌门的普氏菌属-9、链球菌属、黄杆菌属和嗜蛋白菌属高于发酵床夏季样本,分别是夏季样本的3.7、9.6、5.2、3.4、1.9 倍。

图3 微生物发酵床垫料在门水平的微生物组成及相对丰度Figure3 Relative abundances of predominant bacterial compositions on phylum level

图4 微生物发酵床垫料在属水平的微生物组成及相对丰度Figure4 Relative abundances of predominant bacterial compositions on genus level
2.3 微生物发酵床细菌群落季节变化热图分析
通过对不同分类水平的微生物进行多样性分析,选择含量前50的细菌属,根据它们在发酵床夏季和冬季垫料样本中丰度比例结构建立热图(图5)。按季节不同,样本聚成两类:夏季样本(s1、s2和s3)和冬季样本(w1、w2和w3),说明相同季节的微生物发酵床垫料具有相似的微生物构成。根据丰度差异,细菌可聚成3类。第1类为在微生物发酵床夏季和冬季样本中含量低的细菌,包含葡萄球菌属、短杆菌属和涅斯捷连科氏菌等7个属。第二类为夏季样本含量高的降解菌,包括嗜热菌特吕珀菌属、漠河杆菌属、紫单胞菌科、间孢囊菌科及微球菌目的细菌。第三类为冬季样本含量较高的细菌属,其中可分为3亚类。硫假单胞菌属和糖杆菌属含量最高;第二亚类包含嗜蛋白菌属、海洋杆菌属、不动杆菌属和嗜冷杆菌属等9个属的细菌,多为适冷菌;第三亚类包含假单胞菌属、乳酸杆菌属、黄杆菌属等降解菌。
异常球菌-栖热菌门在夏季发酵床含量高,此门细菌包含异常球菌目和栖热菌目2个目,具有厚的细胞壁结构,能够抵抗严酷环境。在微生物发酵床中分离到的该门细菌有异常球菌目的特吕珀菌属。放线菌门在夏季微生物发酵床中的含量也明显高于冬季样本,为冬季的3.5倍。放线菌是自然界中重要的降解菌,可降解多种有机物。拟杆菌门的漠河杆菌属含量也高于冬季样本。
变形菌门细菌在冬季发酵床中含量高。微生物发酵床的该门细菌主要包括:β-变形菌的寡源杆菌属和γ-变形菌的假单胞菌属、硫假单胞菌属、海洋杆菌属等,其中假单胞菌属和硫假单胞菌含量高于夏季样本。冬季样本中拟杆菌门也具有数量优势,是夏季样本的2倍多。拟杆菌门的嗜蛋白菌属、冬季微菌属和黄杆菌属在冬季发酵床中的含量分别为夏季样本的1.8、2.1、5.2 倍。

图5 细菌在各样本间的分布热图(属水平)Figure5 Hierarchically clustered heatmap of bacterial distribution of different communities from the samples on the genus level
2.4 微生物发酵床细菌群落季节变化关联分析
RDA法分析结果反映了垫料菌群与季节性温度之间的关系。结果(图6)显示,细菌门的分布与温度相关。分析结果可以看出:①温度与放线菌门含量夹角为锐角,呈正相关,随温度升高而增加,在夏季发酵床含量高达19.8%,而冬季含量则为12.7%;②温度与厚壁菌门、变形菌门和拟杆菌门的含量为钝角,呈负相关,3个门细菌含量随温度降低而增加,在冬季样本中含量较高,分别是夏季样本的1.4、2.1、1.1倍;③放线菌与厚壁菌门、变形菌门和拟杆菌门菌量之间为钝角,呈负相关,随着温度升高放线菌含量增加,夏季发酵床中的放线菌含量是冬季垫料的1.6倍,而厚壁菌门、变形菌门和拟杆菌门含量分别降低了27.3%、53.4%和10.4%;④厚壁菌门、变形菌门和拟杆菌门之间呈锐角,呈正相关,它们在夏季和冬季发酵垫料中的含量变化一致。
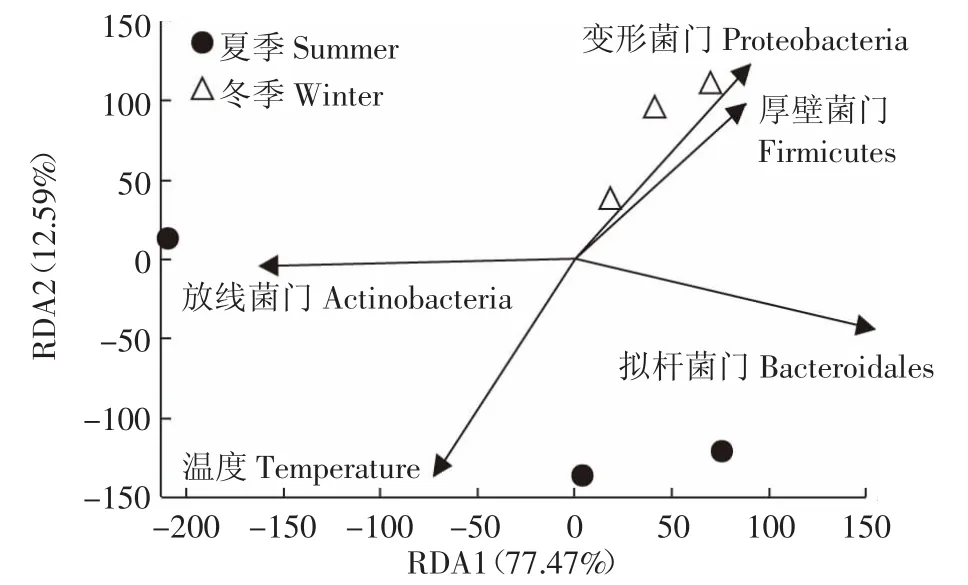
图6 垫料微生物与季节因子的冗余分析Figure6 Redundancy analysis(RDA)of the relationship between bacterial phylum and the season parameters
2.5 发酵床微生物代谢相关基因的季节性差异
根据PICRUSt预测两个季节主要有机物降解途径,分析细菌群落对氨基酸、碳水化合物和脂类代谢的相关基因拷贝数的季节差异性。研究发现夏季高温条件代谢相关基因丰度高,有机物代谢活跃(图7)。夏季发酵床垫料细菌氨基酸、碳水化合物和脂类代谢基因的拷贝数分别为6808477、6480413和2121815,比冬季高27.3%、27.5%和27.1%。如,夏季垫料细菌群落中参与氨基酸代谢的氨基转移酶、肽酶及脱羧酶的拷贝数分别为14 327、58 036和17 157,是冬季的1.5、1.3、1.6倍;糖酵解途径中的葡萄糖激酶、丙酮酸激酶和乳酸脱氢酶拷贝数是8048、28 506及4252,为冬季的1.8、1.1、1.3倍;脂类代谢中的羧酸酯酶和甘油磷酸二酯酶是16 860和44 061,为冬季的2.0、1.5倍。根据PICRUSt的预测信息,夏季发酵床细菌的有机物代谢基因数量高于冬季,推测夏季发酵床垫料细菌的代谢水平高于冬季,与细菌群落丰度和多样性指数高有关。

图7 发酵床细菌群落氨基酸、碳水化合物和脂类代谢相关基因的季节性差异Figure7 Functional prediction of bacterial community in amino acid,carbohydrate and lipid metabolism
3 讨论
畜牧业产生的粪污及其副产物造成的环境污染是制约我国禽畜养殖健康发展的一大障碍。规模化养殖导致大量的粪便集中产生,未经处理排放至环境,会造成水源、土壤和空气等严重污染[20-21]。采用微生物发酵的方式处理禽畜粪便,将养殖废弃物转化为生产有机肥,可实现资源充分利用。本研究分析了微生物发酵床夏季和冬季垫料的细菌多样性,发现微生物发酵床的主要细菌为拟杆菌门、厚壁菌门、变形菌门和放线菌门。李志宇[22]采用传统平板分离培养方法以及PCR-DGGE分子技术分析了发酵床垫料微生物的多样性,发现主要的菌群为厚壁菌门、拟杆菌门和变形菌门。朱双红[23]采用16S rRNA克隆结合酶切分型的方式获得生物发酵床样本中的细菌组成,主要为厚壁菌门、变形菌门和拟杆菌门,这与本研究结果一致。采用宏基因组方法研究微生物发酵床中的细菌组成发现拟杆菌门占主要优势,约为细菌总量的30%,而在李志宇[22]和朱双红[23]的研究中厚壁菌门是主要细菌,占细菌总量的55%以上。由于分子克隆方式通量不够,不能全面揭示环境微生物多样性,这是造成本研究结果与前人研究差异的主要原因,也说明宏基因组技术在环境微生物多样性研究方面具有明显优势。微生物发酵床细菌群落多样性分析结果表明微生物发酵床含有丰富的细菌资源,这为进一步挖掘新物种和新功能提供了重要来源。
夏季和冬季发酵床室温有所差异,同时夏季和冬季发酵床垫料的细菌群落构成也不同。夏季优势菌为特吕珀菌属和漠河菌属;冬季主要为假单胞菌属和硫假单胞菌。特吕珀菌属能够适应高温环境,最佳生长温度为50℃,可利用多种糖类、有机酸和氨基酸[24],在温度较高的夏季(平均室温为30.13℃)相对含量高。漠河杆菌属于拟杆菌门黄杆菌科,分离于粪便发酵物[25],参与粪便的生物降解。夏季高温下,含量具有显著差异的特吕珀菌属和漠河杆菌属参与高温下的粪污降解。冬季发酵床中,假单胞菌属和硫假单胞菌属含量明显高于夏季。假单胞菌分布广泛,是重要的有机物降解菌[26-27]。分离自厌氧发酵的活性污泥中的硫假单胞菌是厌氧菌,可以硝酸盐为电子受体氧化还原活性污泥中的硫化物,参与氮循环过程[28-29]。微生物发酵床中的假单胞菌属和硫假单胞菌属细菌可能与低温下氮的利用相关。此外,在冬季的垫料样本中,嗜冷菌属和冬季微菌属含量也高于夏季样本。嗜冷菌属是变形菌门莫拉氏菌科好寒或耐寒的细菌,常分布于潮湿、冷盐的环境中,在温暖和低盐的环境中也有分布,在食品发酵中起重要作用[30]。冬季微菌属是适冷性细菌,最适生长温度为16~19℃[31],在冬季发酵床平均室温约为16℃,适合生长。冬季微生物发酵床适冷菌和嗜冷菌含量高,与冬季发酵床较低的温度相关。综上,温度是影响发酵床垫料中细菌多样性的重要因素之一。
微生物能够通过有氧或厌氧发酵降解粪污中的有机物并产生能量[32],目前已经有多种微生物和复合微生物菌剂应用于养殖废弃物处理中。肖翰等[33]将微生物菌剂接种至发酵床垫料中,发现接种菌剂能够显著提高微生物数量、发酵温度以及各种酶的活性。王义祥等[34]将复合EM菌剂添加至堆肥发酵系统,促进了有机质的矿化分解和提高腐殖化指数。复合微生物菌剂是一种多菌种共存的生物体系,每种微生物有各自特定的生活环境,许多微生物性能需在特定的环境中表达。因此,菌剂环境的适应性研究是复合微生物菌剂开发中的关键。垫料中的细菌包括能在较高温度下起降解作用的嗜热菌特吕珀菌属、漠河杆菌属、紫单胞菌科、间孢囊菌科及微球菌目的细菌,也包含低温降解菌如假单胞菌属、乳酸杆菌属、黄杆菌属等。微生物发酵床中的这些细菌表现出对高温和低温环境的适应性,为降解养殖废弃物复合微生物菌剂的研发提供基础。
本研究揭示了夏、冬季节发酵床细菌的多样性及其分布特征的差异性:夏季微生物发酵床中的放线菌和嗜热菌等有机质降解菌含量高,促进夏季高温条件下的猪粪降解;在冬季低温条件下嗜冷菌和适冷菌含量高,促进猪粪在较低温度下的降解。对夏、冬季节细菌多样性的分析有助于研究细菌与环境的协同适应,发掘发酵床中蕴藏的耐热、嗜冷和嗜盐等嗜极细菌资源;同时为夏、冬季节发酵床的维护提供理论依据,促进以发酵床设施处理养殖废弃物技术的发展。
4 结论
本研究揭示了夏、冬季节发酵床垫料细菌群落构成,优势菌为拟杆菌门、厚壁菌门、变形菌门和放线菌门。夏、冬季节垫料细菌群落结构不同,前者有更为丰富的细菌类群。夏季样本中的放线菌门和异常球菌-栖热菌门的含量高于冬季样本;异常球菌-栖热菌门的特吕珀菌属含量高于冬季样本。冬季样本中的拟杆菌门和变形菌门含量高于夏季样本;拟杆菌门的嗜蛋白菌属、冬季微菌属和黄杆菌属以及变形菌门的假单胞菌属、硫假单胞菌属和嗜冷菌属含量高于夏季垫料。季节性温度影响微生物发酵床细菌群落结构与代谢水平。
[1]张履祥.沈氏农书[M].北京:中华书局,1956:50.
ZHANG Lü-xiang.Shen Shi Nong Shu[M].Beijing:Zhonghua Book Company,1956:50.
[2]Gadd J.Unnel housing of pigs in livestock environment Ⅳ[C].Fourth International Symposium.American Society of Agricultural Enginneers,1993:1040-1048.
[3]Connor M L.Update on alternative housing systems for pigs[J].Manitoba Swine Seminar Proceedings,1995(8):93-96.
[4]Chan D K O,Chaw D,Lo C Y.Development of an environmentally friendly and cost effective system for the treatment of waste in pig farming[J].Journal of Agricultural Engineering Research,1995,3:11-17.
[5]Deininger A,Tamm M,Krause R,et al.Penetration resistance and water-holding capacity of differently conditioned straw for deep litter housing systems[J].Journal of Agricultural Engineering Research,2000,77(3):335-342.
[6]Kapuinen P.Structures and environment:Deep litter systems for beef cattle housed in uninsulated barns[J].Journal of Agricultural Engineering Research,2001,80(1):87-97.
[7]赵国华,方雅恒,陈 贵.生物发酵床养猪垫料中营养成分和微生物群落研究[J].安徽农业科学,2015,43(8):98-99.
ZHAO Guo-hua,FANG Ya-heng,CHEN Gui.Study on the nutritional components and microbial community in the beddings of pig raising by bio-fermentation bed[J].Journal of Anhui Agricultural Sciences,2015,43(8):98-99.
[8]王 震,尹红梅,刘 标,等.发酵床垫料中优势细菌的分离鉴定及生物学特性研究[J].浙江农业学报,2015,27(1):87-91.
WANG Zhen,YIN Hong-mei,LIU Biao,et al.Biological properties and identification of superior bacteria in deep-litter system for pig breeding[J].Acta Agriculturae Zhejiangensis,2015,27(1):87-91.
[9]刘 波,郑雪芳,朱昌雄,等.脂肪酸生物标记法研究零排放猪舍基质垫层微生物群落多样性[J].生态学报,2008,28(11):5488-5498.
LIU Bo,ZHENG Xue-fang,ZHU Chang-xiong,et al.The diversity of PLFAs biomarkers for the microbial community in the stroma cushion of non-pollution pigsty[J].Acta Ecologica Sinica,2008,28(11):5488-5498.
[10]宦海琳,闫俊书,周维仁,等.不同垫料组成对猪用发酵床细菌群落的影响[J].农业环境科学学报,2014,33(9):1843-1848.
HUAN Hai-lin,YAN Jun-shu,ZHOU Wei-ren,et al.Effects of different bedding litters on bacterial community in pig biobed[J].Journal of Agro-Environment Science,2014,33(9):1843-1848.
[11]李 翔,秦 岭,戴世鲲,等.海洋微生物宏基因组工程进展与展望[J].微生物学报,2007,47(3):548-553.
LI Xiang,QIN Ling,DAI Shi-kun,et al.Marine microbial metagenomics:Progress and prospect[J].Acta Microbiologica Sinica,2007,47(3):548-553.
[12]Ren G M,Xu X H,Qu J J,et al.Evaluation of microbial population dynamics in the co-composting of cow manure and rice straw using high throughput sequencing analysis[J].World Journal of Microbiology and Biotechnology,2016,32(6):101.
[13]Chen Q Q,Liu B,Wang J P,et al.Diversity and dynamics of the bacterial community involved in pig manure biodegradation in a microbial fermentation bed system[J].Annals of Microbiology,2017:67(7):491-500.
[14]Schloss P D,Westcott S L,Ryabin T,et al.Introducing mothur:Opensource,platform-independent,community-supported software for describing and comparing microbial communities[J].Applied Environmental Microbiology,2009,75(23):7537-7541.
[15]Wang Q,Garrity G M,Tiedje J M,et al.Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J].Applied and Environmental Microbiology,2007,73(16):5261-5267.
[16]Quast C,Pruesse E,Yilmaz P,et al.The SILVA ribosomal RNA gene database project:Improved data processing and web-based tools[J].Nucleic Acids Research,2013,41(D1):D590-D596.
[17]Oberauner L,Zachow C,Lackner S,et al.The ignored diversity:Complex bacterial communities in intensive care units revealed by 16S pyrosequencing[J].Scientific Reports,2013,3(3):1413.
[18]Derrick E,Sebastian S,Janaki P,et al.Next generation sequencing to define prokaryotic and fungal diversity in the bovine rumen[J].PLoS One,2012,7(11):e48289.
[19]Langille M G I,Zaneveld J,Caporaso J G,et al.Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences[J].Nature Biotechnology,2013,31(9):814-821.
[20]苏 杨.我国集约化畜禽养殖场污染问题研究[J].中国生态农业学报,2006,14(2):15-18.
SU Yang.Research of countermeasures on waste treating of intensive livestock and poultry farms in China[J].Chinese Journal of Eco-Agriculture,2006,14(2):15-18.
[21]Leconte M C,Mazzarino M J,Satti P,et al.Nitrogen and phosphorus release from poultry manure composts:The role of carbonaceous bulking agents and compost particle sizes[J].Biology and Fertility of Soils,2011,47(8):897-906.
[22]李志宇.动物养殖发酵床中微生物变化规律的研究[D].大连:大连理工大学,2012.
LI Zhi-yu.Microbial community variation in fermention-bed system for farming animals[D].Dalian:Dalian University of Technology,2012.
[23]朱双红.猪生物发酵床垫料中细菌群落结构动态变化研究[D].武汉:华中农业大学,2012.
ZHU Shuang-hong.The dynamic changes of bacterial community structure on micro-fermention bed piggery litter[D].Wuhan:Huazhong Agriculture University,2012.
[24]Albuquerque L,Simões C,Nobre M F,et al.Truepera radiovictrix,gen.nov.sp.nov.A new radiation resistant species and the proposal of Trueperaceae.fam.nov.[J].Fems Microbiology Letters,2005,247(2):161-169.
[25]Schauss T,Busse H J,Golke J,et al.Moheibacter stercoris sp.nov isolated from an input sample of a German biogas plant[J].International Journal of Systematic and Evolutionary Microbiology,2016,150(4):1-16.
[26]郝燕妮,林建国,郭 平,等.一株具有石油烃降解性能的交替假单胞菌的筛选和鉴定[J].科学技术与工程,2016,16(5):142-146.
HAO Yan-ni,LIN Jian-guo,GUO Ping,et al.Isolation and identification of Pseudoalteromonas strain with petroleum degrading properties[J].Science Technology and Engineering,2016,16(5):142-146.
[27]李 静,邓毛程,王 瑶,等.共代谢基质促进铜绿假单胞菌降解三十六烷的研究[J].环境科技,2016,29(4):11-14.
LI Jing, DENG Mao-cheng, WANG Yao, et al. Influence of co -metabolism substrates on hexatriacontane biodegradation by Pseu -domonas aeruginosa LJ06[J]. Environmental Science and Technology,2016, 29(4):11-14.
[28]Tan W B, Huang C, Chen C, et al. Bioaugmentation of activated sludgewith elemental sulfur producing strain Thiopseudomonas denitrificans,X2 against nitrate shock load[J]. Bioresource Technology, 2016, 220:647-650.
[29]Tan W B,Jiang Z,Chen C,et al.Thiopseudomonas denitrificans gen.nov.,sp.nov.,isolated from anaerobic activated sludge[J].International Journal of Systematic and Evolutionary Microbiology,2015,65(Pt1):225-238.
[30]Juni E,Heym G A.Psychrobacter immobilis gen.nov.,sp.nov.:Genospecies composed of gram-negative,aerobic,oxidase-positive coccobacilli[J].International Journal of Systematic Bacteriology,1986,36(3):388-391.
[31]Bowman J P, Nichols C M, Gibson J A. Algoriphagus ratkowskyi gen.nov., sp. nov., Brumimicrobium glaciale gen. nov., sp. nov., Cryomorphaignava gen. nov., sp. nov. and Crocinitomix catalasitica gen. nov., sp.nov., novel flavobacteria isolated from various polar habitats[J]. InternationalJournal of Systematic and Evolutionary Microbiology, 2003, 53(Pt5):1343-1355.
[32]陈 杰,赵祥杰,邝哲师,等.利用微生物处理畜禽粪便的研究[J].安徽农业科学,2014,42(28):9910-9911,9944.
CHEN Jie,ZHAO Xiang-jie,KUANG Zhe-shi,et al.Study on animal slurry treatment by microorganism[J].Journal of Anhui Agricultural Sciences,2014,42(28):9910-9911,9944.
[33]肖 翰,雷 平,刘 标,等.接种菌剂对发酵床垫料中微生物数量与酶活性的影响[J].江苏农业科学,2017,45(8):157-159.
XIAO Han,LEI Ping,LIU Biao,et al.Effects of inoculating bacteria on microbial quantity and enzyme activity in fermented bedding[J].Jiangsu Agricultural Sciences,2017,45(8):157-159.
[34]王义祥,高凌飞,叶 菁,等.菌渣垫料堆肥过程碳素物质转化规律[J].农业工程学报,2016,32(增刊 2):292-296.
WANG Yi-xiang,GAO Ling-fei,YE Jing,et al.Change of carbon substance characteristics during composting of waste packing and fungus chaff[J].Transactions of the Chinese Society of Agricultural Engineering,2016,32(Suppl 2):292-296.
