通过三组分串联反应构建硫醚化吲哚衍生物
2018-06-27苏智扬李博洋刘正浩占赛松申红梅史亚男
苏智扬 ,李博洋,刘正浩 ,占赛松 ,申红梅 ,史亚男 ,赵 霞
(1.天津师范大学化学学院,天津300387;2.天津师范大学无机-有机杂化功能材料化学教育部重点实验室,天津300387;3.天津师范大学天津市功能分子结构与性能重点实验室,天津300387)
有机硫化合物是许多药物分子或材料分子的重要片段或重要结构单元[1],在有机硫化合物的合成反应中,碳-硫键的形成起到了非常关键的作用.因此,近年来对碳-硫键构建新方法的探索备受关注[2].以往碳-硫键的形成反应由过渡金属催化,虽可避免恶劣的操作条件,如高沸点的有毒极性溶剂,但催化剂价格昂贵且对空气敏感,再加上硫源的不稳定和挥发性,限制了此类反应的实际应用[3-4].作为非金属催化剂,相比过渡金属催化剂,碘具有廉价、对空气不敏感等特点,因此已广泛应用于碳-碳键、碳-氮键、碳-硫键的构建反应中.如相李奎[5]利用碘作催化剂,实现了多取代吲嗪和咪唑化合物的合成.Chen等[6]以碘为催化剂,利用二硫化物或二硒化物在吲哚骨架上构建了碳-硫键、碳-硒键.除了对催化剂的改良外,化学工作者也在不断探索硫醚化试剂.近年来出现了一种新型环保的硫醚化试剂,即磺酰肼,该类化合物具有化学性质稳定、无异味等优点,被用于碳-硫键的形成反应.如Yang等[7]以苯磺酰肼类化合物为硫醚化试剂,以碘为催化剂,在吲哚骨架上构建了碳-硫键.磺酰氯作为为磺酰肼合成的前体,以其为硫醚化试剂构建碳-硫键的反应也得到了发展.如Wu等[8]于2011年首次利用苯磺酰氯类化合物作为硫醚化试剂,利用三苯基膦作催化还原剂,于130℃条件下成功地在吲哚嗪、吲哚上构建了碳-硫键;Chen等[9]以芳基磺酰氯为硫醚化试剂,利用钌配合物作催化剂,在光照条件下,实现了N-甲基吲哚上的碳-硫键构建反应.本课题组前期也以磺酰肼或磺酰氯作为硫醚化试剂,发展了多种碳-硫键的形成反应[10-13].
本研究利用三组分串联反应,在碘的催化作用下,将对甲苯磺酰氯或其衍生物、水合肼与吲哚类化合物混合,发生一步反应,直接在吲哚类化合物上构建了碳-硫键.
1 材料与方法
1.1 仪器与试剂
仪器:核磁共振光谱(1H NMR)测试仪,德国Bruker公司,以四甲基硅烷(TMS)为内标.
试剂:乙腈、1,4-二氧六环、N,N-二甲基甲酰胺、1,2-二氯乙烷,天津基准化学试剂有限公司.所有试剂均为分析纯级.
1.2 实验步骤及反应条件优化
对甲苯磺酰氯、水合肼与吲哚反应生成吲哚芳基硫醚的方程式如图1所示.
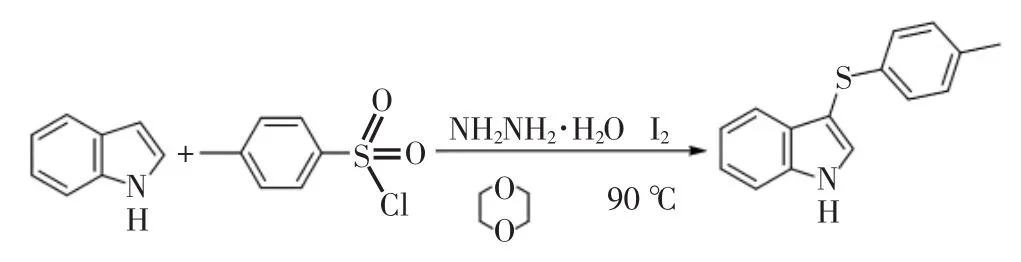
图1 对甲苯磺酰氯、水合肼与吲哚反应的方程式Fig.1 Chemical equation of p-toluene-sulfonyl chloride,hydrazine hydrate and indole
对甲苯磺酰氯、水合肼与吲哚反应的通用步骤:将吲哚和对甲苯磺酰氯加入到15 mL耐压瓶中,然后依次加入一定量的溶剂、水合肼和碘单质,充分搅拌反应.反应一段时间后,用薄层色谱法检测,当发现薄层色谱板上原料点紫外显色变淡直至无明显变化时,视为反应完毕,停止加热.将体系降至室温后转移至10 mL圆底烧瓶,减压蒸发旋干溶剂,得到待过柱分离物.用硅胶-柱色谱将产物分离出来,洗脱剂为石油醚和乙酸乙酯混合物(体积比为10∶1).
反应体系中,吲哚固定为0.5 mmol,分别选用水、甲苯(toluene)、乙腈(MeCN)、乙醇(EtOH)、二甲基甲酰胺(DMF)、1,4-二氧六环(1,4-dioxane)、1,2-二氯乙烷(DCE)为溶剂.对溶剂及其用量、反应温度、对甲苯磺酰氯和水合肼的用量、碘单质用量进行筛选,以吲哚芳基硫醚产率最高时的反应条件为最优.
1.3 实验底物的扩展
分别选用连有不同取代基团的吲哚和连有不同取代基团的苯磺酰氯,在水合肼存在下,使用碘单质作为催化剂,在1.2中得到的最优反应条件下进行三组分串联反应,得到多种硫醚化吲哚衍生物.反应通式如图2所示.
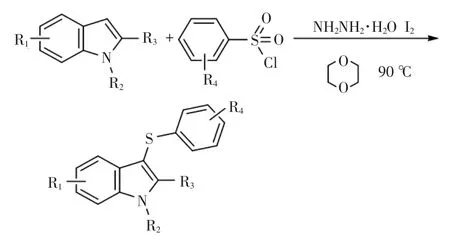
图2 底物扩展实验的反应通式Fig.2 General reaction formula of substrate expansion experiments
2 结果与分析
2.1 对甲苯磺酰氯、水合肼与吲哚串联反应条件的筛选
反应体系中,吲哚固定为0.5 mmol,对溶剂的种类和用量、反应温度、对甲苯磺酰氯和水合肼的用量、碘单质用量进行筛选,不同反应条件的设置和吲哚芳基硫醚的产率如表1所示.由表1可以看出,三组分串联反应的最优反应条件为:吲哚(0.5 mmol)、对甲苯磺酰氯(0.50 mmol)、水合肼(1.0mmol)、I2(0.050mmol)、溶剂1,4-二氧六环(0.5mL)、反应温度 90℃.最终得到淡黄色的吲哚芳基硫醚固体产物81.7 mg,产率为68%.1H NMR(400 MHz,CDCl3):δ 8.36(s,1H),7.61(dd,J=7.9,1.1 Hz,1H),7.48(d,J=2.6 Hz,1H),7.43(d,J=8.0 Hz,1H),7.29~7.24(m,1H),7.15(td,J=7.5,7.0,1.0Hz,1H),7.06~7.00(m,2H),6.97(d,J=8.1 Hz,2H),2.24(s,3H).
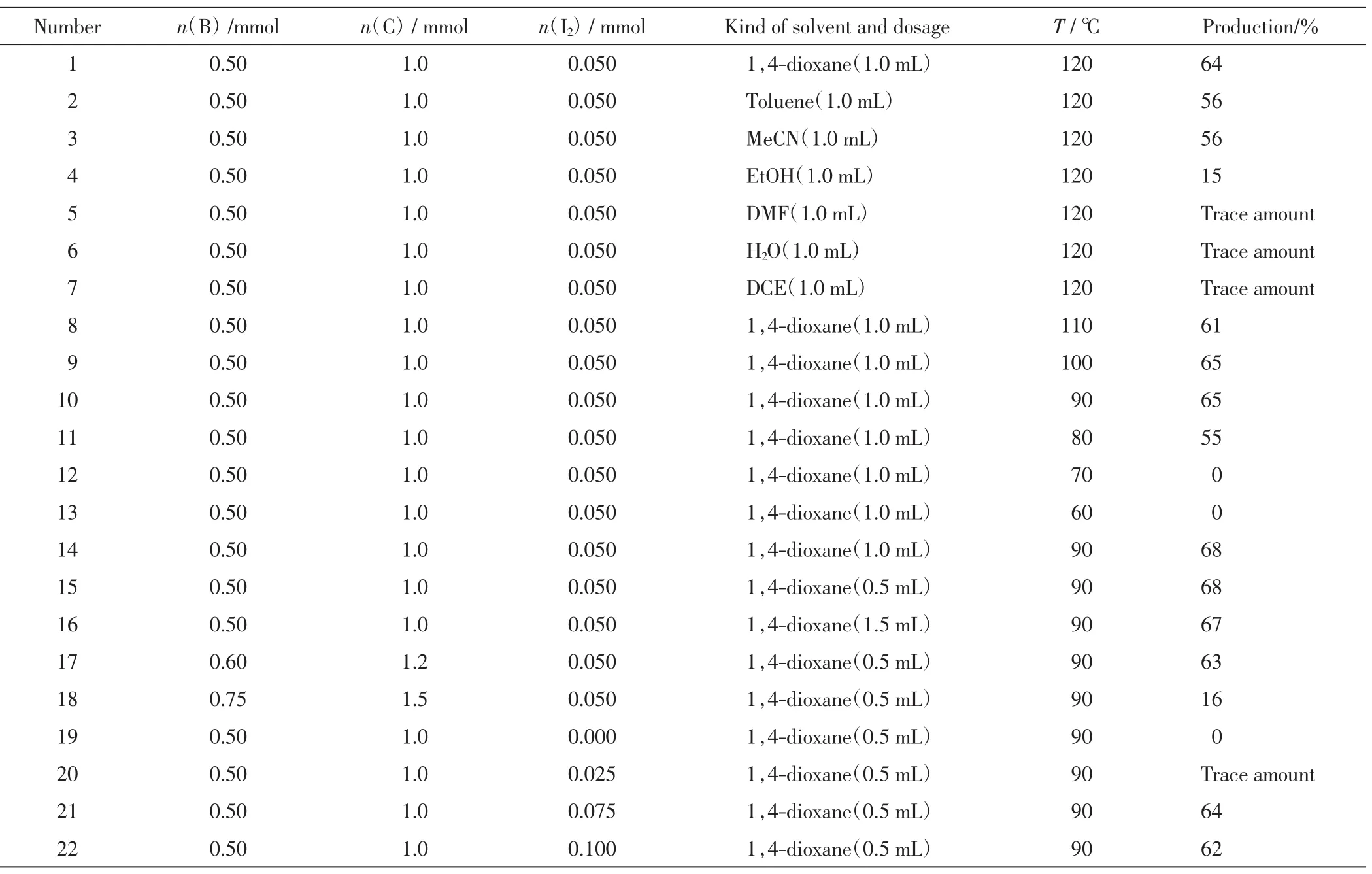
表1 三组分串联反应的条件筛选Tab.1 Conditional screening of three components cascade reaction
2.2 碘单质催化的对甲苯磺酰氯、水合肼与吲哚串联反应底物的适用性
在2.1中得到的最优条件下,选用连有不同取代基团的吲哚参与反应,得到12种化合物;选取连有不同取代基团的苯磺酰氯参与反应,得到2种化合物.吲哚芳基物硫醚(a)和 14种衍生物(b~o)的结构式如图3所示,取代基和产率如表2所示.

图3 吲哚芳基硫醚及衍生物的结构式Fig.3 Formulas of indole aryl thioether and its derivatives
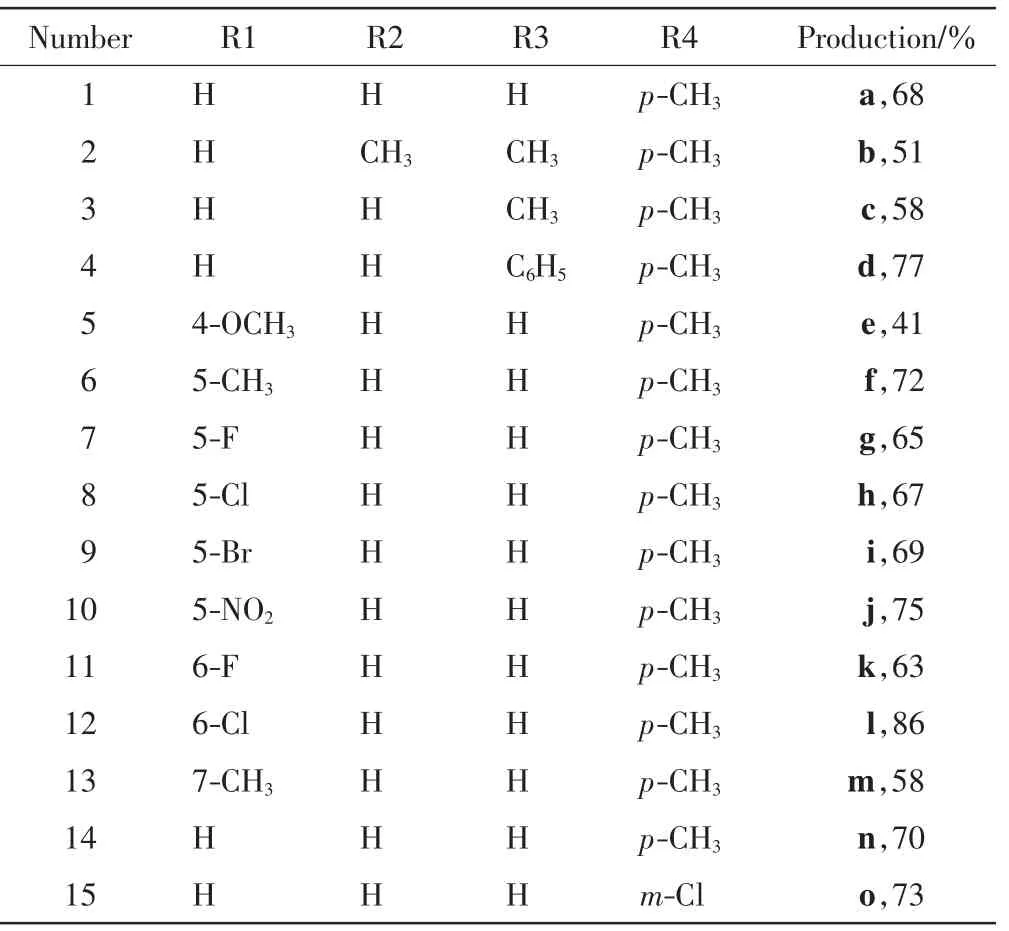
表2 底物扩展实验的产率Tab.2 Production of substrate extension experiments and production
从表2可以看出,连有供电子基团的吲哚参与反应可获得中等收率,如2-甲基吲哚、5-甲基吲哚和7-甲基吲哚,产物分别为c、f、m;当吲哚上连有吸电子基团时,其参与的反应也可获得中等至较高收率,如5-氯吲哚、5-溴吲哚、5-硝基吲哚、6-氯吲哚,对应产物分别为h、i、j、l.吲哚衍生物的选取除了根据基团的电子效应外,还可根据吲哚上不同的取代位点对取代基团进行选择.从表2中可以看出,吲哚的1,2-二取代、5-取代、6-取代、7-取代衍生物参与的反应都可获得中等至较高收率.存在空间位阻的2-甲基苯磺酰氯参与的反应也可获得较高收率(70%).
化合物a:淡黄色固体(81.7 mg,68%).1H NMR(400 MHz,CDCl3)δ 8.36(s,1H),7.61(dd,J=7.9,1.1 Hz,1H),7.48 (d,J=2.6 Hz,1H),7.43 (d,J=8.0 Hz,1H),7.29 ~ 7.22(m,1H),7.15(td,J=7.5,7.0,1.0 Hz,1H),7.06 ~ 7.00(m,2H),6.97(d,J=8.1 Hz,2H),2.24(s,3H).
化合物b:黄色固体(68.1 mg,51%).1H NMR(400 MHz,CDCl3)δ 7.57 (d,J=7.8 Hz,1H),7.31(s,1H),7.20(t,J=7.6 Hz,1H),7.11(t,J=7.4 Hz,1H),6.93(s,4H),3.71(s,3H),2.49(s,3H),2.22(s,3H).
化合物c:淡灰粉色固体(73.2 mg,58%).1H NMR(400 MHz,CDCl3)δ 8.21(s,1H),7.54(d,J=7.7 Hz,1H),7.34 (d,J=8.0Hz,1H),7.21~7.15 (m,1H),7.15 ~7.09(m,1H),6.96(s,4H),2.52(s,3H),2.24(s,3H).
化合物d:褐色固体(121.2 mg,77%).1H NMR(400 MHz,CDCl3)δ 8.40(s,1H),7.71(d,J=7.4 Hz,1H),7.62(d,J=7.8 Hz,1H),7.44~ 7.30(m,5H),7.24~ 7.17(m,1H),7.14(t,J=7.4 Hz,1H),7.01 ~6.93(m,4H),2.21(s,3H).
化合物e:淡黄色固体(54.9 mg,41%).1H NMR(400 MHz,CDCl3)δ 8.30(s,1H),7.18(d,J=2.0 Hz,1H),7.13 (d,J=8.0Hz,1H),7.10 (d,J=1.8Hz,1H),7.09(s,1H),6.99(s,1H),6.97(s,1H),6.95(s,1H),6.52(d,J=7.8 Hz,1H),3.71(s,3H),2.24(s,3H).
化合物f:淡粉色固体(91.0 mg,72%).1H NMR(400 MHz,CDCl3)δ 8.28(s,1H),7.43(d,J=2.5 Hz,1H),7.41(s,1H),7.32(d,J=8.3 Hz,1H),7.08(d,J=8.0Hz,1H),7.00 (q,J=8.2Hz,4H),2.41 (s,3H),2.25(s,3H).
化合物 g:黄色固体(83.3 mg,65%).1H NMR(400MHz,CDCl3)δ 8.33(s,1H),7.48(s,1H),7.32(dd,J=8.7,3.8 Hz,1H),7.24(s,1H),7.03(s,1H),7.00(d,J=7.1 Hz,4H),2.25(s,3H).
化合物h:土黄色固体(92.0 mg,67%).1H NMR(400 MHz,CDCl3)δ 8.36(s,1H),7.58(s,1H),7.45(s,1H),7.31(d,J=8.6 Hz,1H),7.19(d,J=8.6 Hz,1H),7.01(m,4H),2.25(s,3H).
化合物i:土黄色固体(109.4 mg,69%).1H NMR(400MHz,CDCl3)δ8.37(s,1H),7.75(s,1H),7.44(s,1H),7.33(d,J=8.6 Hz,1H),7.27(d,J=8.6 Hz,1H),7.00(m,4H),2.25(s,3H).
化合物j:橙黄色固体(105.9 mg,75%).1H NMR(400 MHz,CDCl3)δ 8.99(s,1H),8.56(s,1H),8.15(d,J=8.9 Hz,1H),7.64(s,1H),7.48(d,J=9.0 Hz,1H),7.07~6.98(m,4H),2.26(s,3H).
化合物k:淡黄色固体(80.6 mg,63%).1H NMR(400 MHz,CDCl3)δ 8.30(s,1H),7.49(dd,J=8.6,5.4Hz,1H),7.41(d,J=2.1Hz,1H),7.08(d,J=9.3 Hz,1H),7.00 (q,J=8.2Hz,4H),6.90 (t,J=9.1Hz,1H),2.25(s,3H).
化合物l:淡黄色固体(117.3 mg,86%).1H NMR(400 MHz,CDCl3)δ 8.32(s,1H),7.49(d,J=8.5 Hz,1H),7.46~ 7.43(m,1H),7.40(s,1H),7.11(d,J=8.4 Hz,1H),7.02~ 6.97(m,4H),2.25(s,3H).
化合物m:黄色固体(73.2 mg,58%).1H NMR(400 MHz,CDCl3)δ 8.30(s,1H),7.48(d,J=2.7 Hz,1H),7.47~ 7.43(m,1H),7.05(dd,J=17.4,7.6 Hz,4H),6.96(d,J=8.1 Hz,2H),2.53(s,3H),2.24(s,3H).
化合物n:淡粉色固体(83.6 mg,70%).1H NMR(400 MHz,CDCl3)δ 8.42(s,1H),7.58(d,J=7.9 Hz,1H),7.48(d,J=2.6 Hz,1H),7.45(d,J=8.2 Hz,1H),7.28 (d,J=7.3 Hz,1H),7.17 (d,J=7.3 Hz,1H),7.15~ 7.11(m,1H),7.00~ 6.94(m,1H),6.89(t,J=7.5 Hz,1H),6.71(d,J=7.8 Hz,1H),2.49(s,3H).
化合物o:淡黄色固体(94.3 mg,73%).1H NMR(400 MHz,CDCl3)δ 8.45(s,1H),7.59(d,J=7.9 Hz,1H),7.50 (d,J=2.6 Hz,1H),7.46 (d,J=8.2 Hz,1H),7.31~7.26(m,1H),7.21~7.16(m,1H),7.08(d,J=7.8 Hz,1H),7.05(d,J=1.8 Hz,1H),7.01(dt,J=7.7,1.5 Hz,1H),6.97(dt,J=7.6,1.5 Hz,1H).
所有化合物的核磁氢谱数据与文献[14-19]的数据相吻合,其中,2-甲基吲哚硫醚化衍生物(c)、2-苯基吲哚硫醚化衍生物(d)、5-甲基吲哚硫醚化衍生物(f)、5-溴吲哚硫醚化衍生物(i)和6-氯吲哚硫醚化衍生物(l)的收率远远大于其他研究中相同产物的收率[15,17-20].即使是连有强吸电子基团的5-硝基吲哚,采用该方法进行硫醚化,获得的5-硝基吲哚硫醚化衍生物(j)的收率也比其他一些用硫醚化方法获得的5-硝基吲哚硫醚化产物的收率要高得多[8,21].
3 结论
本研究利用芳基磺酰氯和水合肼原位生成磺酰肼,在碘的催化下磺酰肼分解并与吲哚类化合物构建了碳-硫键.最优反应条件为:吲哚(0.5 mmol)、对甲苯磺酰氯(0.50 mmol)、水合肼(1.0 mmol)、I2(0.050 mmol)、溶剂 1,4-二氧六环(0.5 mL)、反应温度 90 ℃.在此条件下,选取连有不同取代基团的吲哚类化合物进行硫醚化反应,得到了吲哚芳基硫醚及14种硫醚化吲哚衍生物,产率最高可达86%.该反应底物适用性良好,反应条件温和,所用催化剂为非金属催化剂.此外,采用三组分串联法对吲哚类化合物进行碳-硫键构建,同采用两步法合成硫醚化吲哚相比,合成步骤缩短,反应效率及反应原子的利用率提高.用该合成方法获得的硫醚化吲哚衍生物也可以得到较高收率.
[1]KONDO T,MITSUDO T A.Metal-catalyzed carbon-sulfur bond formation[J].Chem Rev,2000,100(8):3205-3220.
[2]ZHANGXY,ZENG W L,YANG Y,et al.Copper-catalyzed double C-S bonds formation via different paths:Synthesis of benzothiazoles from N-benzyl-2-iodoaniline and potassium sulfide[J].Org Lett,2014,16(3):876-879.
[3]FERNÁNDEZ-RODRÍGUEZ M A,HARTWIG J F.One-pot synthesis of unsymmetrical diaryl thioethers by palladium-catalyzed coupling of two aryl bromides and a thiol surrogate[J].Chem Eur J,2010,16(8):2355-2359.
[4]ZHANG S H,QIAN P C,ZHANG M L,et al.Copper-catalyzed thiolation of the di-or trimethoxybenzene arene C-H bond with disulfides[J].J Org Chem,2010,75(19):6732-6735.
[5]相李奎.碘催化多取代吲嗪和咪唑合成方法的研究[D].兰州:兰州大学,2016 XIANG L K.I2-Catalyzed Synthesis of Substituted Indolizines and Imidazoles[D].Lanzhou:Lanzhou University,2016(in Chinese).
[6]CHEN S Q,WANG Q M,XU P C,et al.Iodine-promoted selective 3-selanylation and 3-sulfenylation of indoles with dichalcogenides under mild conditions[J].Phosphorus&Sulfur&the Related Elements,2016,191(1):100-103.
[7]YANG F L,TIAN S K.Iodine-catalyzed regioselective sulfenylation of indoleswithsulfonylhydrazides[J].AngewChemIntEd,2013,52(18):4929-4932.
[8]WU Q,ZhAO D B,QIN X R,et al.Synthesis of di(hetero)aryl sulfides by directly using arylsulfonyl chlorides as a sulfur source[J].Chem Commun,2011,47(32):9188-9190.
[9]CHEN M,HUANG Z T,ZHENG Q Y.Visible light-induced 3-sulfenylation of N-methylindoles with arylsulfonyl chlorides[J].Chem Commun,2012,48(95):11686-11688.
[10]ZHAO X,ZHANG L P,LI T J,et al.p-Toluenesulphonic acid-promoted,I2-catalysed sulphenylation of pyrazolones with aryl sulphonyl hydrazides[J].Chem Commun,2014,50(86):13121-13123.
[11]ZHAO X,ZHANG L P,LU X Y,et al.Synthesis of 2-aryl and 3-aryl benzo[b]furan thioethers using aryl sulfonyl hydrazides as sulfenylation reagents[J].J Org Chem,2015,80(5):2918-2924.
[12]ZHAO X,LI T J,ZHANG L P,et al.Iodine-catalyzed thiolation of electron-richaromaticsusingsulfonylhydrazidesassulfenylationreagents[J].Org Biomol Chem,2016,14(3):1131-1137.
[13]ZHAO X,LU X Y,WEI A Q,et al.Potassium iodide promoted thiolation of pyrazolones and benzofurans using aryl sulfonyl chlorides as sulfenylationreagents[J].TetrahedronLett,2016,57(48):5330-5333.
[14]WU Z M,LI Y C,DING W Z,et al.Copper-catalyzed regioselective sulfenylationofindoleswith arylsulfonyl chlorides[J].Asian J Org Chem,2016,5(5):625-628.
[15]RAO H H,WANG P,WANG J C,et al.K2S2O8/Arenesulfinate:An unprecedented thiolating system enabling selective sulfenylation of indoles under metal-free conditions[J].RSC Adv,2014,4(90):49165-49169.
[16]RAHAMAN R,DEVI N,BHAGAWATI J R,et al.Microwave-assisted regioselectivesulfenylationofindolesundersolvent-andmetal-freeconditions[J].RSC Adv,2016,6(23):18929-18935.
[17]LI X W,XU Y L,WU W Q,et al.Copper-catalyzed aerobic oxidative N-S bond functionalization for C-S bond formation:Regio-and stereoselective synthesis of sulfones and thioethers[J].Chem Eur J,2014,20(26):7911-7915.
[18]WANG F X,ZHOU S D,WANG C M,et al.N-hydroxy sulfonamides as new sulfenylating agents for the functionalization of aromatic compounds[J].Org Biomol Chem,2017,15(25):5284-5288.
[19]LIU X X,CUI H H,YANG D S,et al.Iodine-catalyzed direct thiolation of indoles with thiols leading to 3-thioindoles using air as the oxidant[J].Catalysis Lett,2016,146(9):1743-1748.
[20]GUO T,WEI X N.Ammonium iodide-mediated sulfenylation of 4-hydroxycoumarins or 4-hydroxyquinolinones with a sulfonyl chloride as a sulfur source[J].Synlett,2017,28(18):2499-2504.
[21]NOOKARAJU U,BEGARI E,YETRA R R,et al.CeCl3·7H2O-NaI promoted regioselective sulfenylation of indoles with sulfonylhydrazides[J].Chemistry Select,2016 ,1(1):81-85.
