医疗机构中药制剂的研究
——孙冬梅教授学术经验撷要
2018-06-26
广东省第二中医院(广东省中医药工程技术研究院)/广东省中医药研究开发重点实验室/广州中医药大学第五临床医学院,广东 广州 510095
随着国家加大了药事法规建设的力度,医疗机构中药制剂的法规及其监督机制得以不断发展和完善,相关法规的颁布以及医药市场的变化都将对医疗机构中药制剂的内涵建设、品种及规模产生较大的影响。同时,随着医疗改革的不断深入,中医药事业的发展倍受全社会的广泛关注,国家、地方先后出台了扶持和促进中医药事业发展的若干政策,医疗机构中药制剂迎来了重大的战略机遇期[1]。
中医医疗机构的建设和发展应突显出自身的中医药特色优势,开展精准医疗、个性化、差异化服务。医疗机构中药制剂处方来源于临床经验丰富的中医师,是多年临床用药的经验积累,基于市场上尚无销售,为方便临床方便应用研发而成的药物。
孙冬梅教授自2002年起开始对医疗机构制剂进行规范研发,严格控制医疗机构中药制剂的质量水平,中药制剂的质量直接关系到临床用药的安全性、有效性。中药制剂是多靶点、多成分的作用,影响制剂质量的因素错综复杂。孙教授指出研究医疗机构制剂必须系统、全面,在传承和发展中医药的前提下创新研究方法,运用现代的制药工具、分析方法、独特的评价体系全面研发中药制剂。
1 处方筛选研究
临床处方直接作为中药制剂处方,一般会面临处方药味多、药量大等问题,从而致使制备工艺困难、成品包装过大等,若随意修改处方以减少用药与用量,则药效可能降低。因此,制剂研发首先要做的就是临床处方的筛选,中药复方中单味药用量的配伍有一定比例关系,医师充分论证处方药味与病、证、症的相关性,确定处方药味;药学研究人员可以根据处方中各药味药效成分及其药理毒理作用的相互关系,方中药味的量发生变化时药效也往往随之改变,需对处方药味量比例、日服用量、制剂可行性等进行论证,提出处方修改方案和修改理由,供医师参考;然后在临床上反复验证,使制剂处方用药与用量趋于合理,形成供研发的制剂处方,且有利于进一步的新药开发,即“优选处方-修改处方-临床使用-确定处方”的筛选模式[2]。通过对处方进行筛选,寻找最佳配伍药味及剂量配比关系,增强药物疗效,从而节约生产成本,提高中药的利用率。
我院治疗类风湿性关节炎的制剂,临床处方有11味药,采用人类类风湿关节炎的经典模型,Ⅱ型胶原蛋白动物模型,筛选出由5味药组成的新处方供临床医师进一步验证,经过临床医师的临床试验结果反馈,调整了处方的药味量比例,优化了处方成药性更强。在处方筛选上孙冬梅教授采用药理药效试验进行筛选,筛选的处方再交回临床医师进行临床验证,最终确定适应制剂工艺的最佳处方。
2 制剂工艺研究
中药制剂的工艺研究是质量研究和稳定性研究的基础,是医疗机构制剂研究的重要环节。在制剂工艺中,要根据处方中各药味的有效成分选择合适的工艺路线,研究的重点在提取、浓缩、除杂、干燥、成型等环节,由于中药复方成分复杂,药味间相互影响普遍,因此工艺路线的研究是研发难点之一。
孙冬梅教授强调,在进行制剂工艺研究前必须对药材进行基原鉴定,确保研究的药材来源清晰质量稳定可控,并按《中国药典》的标准要求进行药材检测,符合要求后再进行提取工艺路线的设计,采用正交试验、均匀设计等试验方法进行提取条件研究。在分离、纯化、精制和干燥研究中,孙教授采用现代分析技术对提取物的收得率和相关化学成分的保留率来评价该工艺过程的可行性和合理性。最后根据临床要求、剂型特点、给药途径和提取物的性质进行制剂成型工艺的研究。只有经过系统而又科学的工艺研究,才能确保所研发的中药制剂具有有效、安全、均一和稳定的质量特性,药品质量是否合格,不是检验出来的,而是设计和生产出来的[3]。合理的制剂工艺保证了药物的质量,发挥原有的临床疗效。
孙冬梅教授强调在工艺设计中应考虑药材不同的特性选择不同的工艺方法,例如苍术、薄荷等含有挥发性成份并以挥发性成分起主要药效作用的药材,可考虑单独提取;需特殊处理的贝壳、矿物类药,如鳖甲、磁石等应进行先煎;处方中含有贵细药材,如冰片、血竭等可单独粉碎,原粉入药;含有鲜品的制剂处方可将鲜品单独处理;需要根据处方的实际情况去除杂质,经过纯化的工艺,减少药物的服用量。
例如:我院制剂通阳宽胸颗粒中含有桂枝、陈皮、枳实、川芎四味药材均含有挥发油,挥发油是本方通阳化浊,行气宽胸的重要活性部位,应对其进行挥发油的提取。根据预实验结果,以水蒸气蒸馏法或共水蒸馏法进行提取。①提取时间考察,按处方比例称取桂枝、陈皮、枳实、川芎四味药材共150 g,加入10倍量的水,提取挥发油,记录不同时间的挥发油收油量,考察提取时间对收油率的影响,结果随着时间的延长,挥发油的收集不断增多,提取3 h的提取率约相当于提取5小时提取率的96.4%,为缩短生产周期,降低生产成本等综合因素,选取先进行3 h提取挥发油。②浸泡时间的考察,按处方比例称取桂枝、陈皮、枳实、川芎四味药材共150 g,分别加水10倍量,提取挥发油3小时,其中一份为加水后浸泡过夜,另一份加水后直接回流,考察浸泡过程对挥发油提取结果的影响,结果浸泡与否不影响挥发油的提取率,故本工艺采用药材加水后不浸泡,直接加热回流提取。③加水量的考察,按处方比例称取桂枝、陈皮、枳实、川芎四味药材共150 g,以挥发油提取率为考察指标,分别加水6、8、10倍量,水蒸汽蒸馏提取挥发油3 h,结果加水倍量对挥发油的提取无明显影响,故选取加6倍水提取挥发油。最后对挥发油提取工艺进行验证最终确定挥发油提取工艺参数。
科学合理的选择中药复方的提取工艺是保持原方疗效的关键和核心,也是中药剂型改进和创新的前提和基础。针对处方中药物的性质和现代药理学研究进展,设计合理的、适应实际生产需要的制剂工艺。
3 质量标准研究
中药制剂质量标准是品种注册后的法定文件,是制剂配制、质量控制、临床使用的依据,其内容通常包括制剂名称、处方、制法、性状、鉴别、检查、含量测定、功能主治、用法用量、注意、规格、贮藏、有效期等条目,内容繁多,涉及面广,质量标准研究贯穿整个研发过程。质量控制的难点主要在薄层色谱鉴别和含量测定方法建立,单味药分析往往采用指标成分,而复方制剂存在多成分干扰与掩盖的现象。
医疗机构制剂质量的优劣直接影响到临床用药的安全性和有效性,也关系到患者的健康安全,因此必须建立严格的质量控制标准[4]。孙冬梅教授提出对中药制剂质量的评价和控制,就符合中药作用的整体性、作用靶点和机制的复杂性、组成成分的多样性和可变性,以及运用中医基础理论的整体观,体现中医用药的特色。
孙教授强调制剂中含有粉末入药的药味需要进行显微鉴别研究,如妇宁袋泡茶中的益母草为部分粉末入药,质量标准中采用了显微鉴别,取本粉末,置显微镜下观察,可见腺鳞散在,腺鳞头部4、6细胞或8细胞,直径60~140μm(益母草),如图1所示。对于制剂中含有的主要药味采用薄层色谱法进行鉴别,如祛瘀脉通颗粒中的柴胡、赤芍和白术,见图2~4。针对制剂中主要成分进行浸出物的测定,如三芪丹颗粒,制剂中主要含皂苷类成分,该类成分在水饱和正丁醇中溶解性较好,以水饱和正丁醇为浸出溶剂,采用热浸法测定制剂中的浸出物不得少于10%。
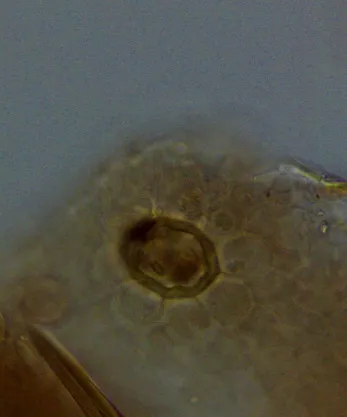
图1 益母草显微鉴别图(腺鳞)
4 稳定性研究
药品生产出来到患者使用,经过贮藏、运输等环节,有可能因为外界的一些因素而变质、含量下降、有效性下降等,一旦有毒性物质产生则影响到药品的安全性。为了保证药物安全有效,对医疗机构中药制剂进行稳定性研究,弄清制剂在不同环境因素影响下质量是否经一定时间后改变,找出影响稳定性的因素,采取相应措施,以阻止或延缓其发生变化,保证药物疗效好、毒副作用小。
药物的稳定性研究包括物理稳定性、微生物稳定性和化学稳定性,其中以化学稳定性较为重要和常见,是药物稳定性研究的主要方面。稳定性研究一般从三个方面进行研究,(1)影响因素试验,在暴露空气中、经强光照射及高湿、高温等环境研究制剂对光、热、湿度和空气等敏感的特性;(2)加速稳定性试验,对制剂在短时间内施加强应力,促使制剂加速发生反应,并按一定的方法,推测其有效期,因大多数药品的反应速率会随湿度升高而显著加快,通常以温度作为强应力,一般是受试制剂在上市包装条件下进行考察,将三批中试产品在(40±2)℃、相对湿度(75±5)%的条件下考察6个月,样品性状、鉴别、检查等各项指标均无明显变化,说明该制剂质量稳定;(3)长期稳定性试验,目的是考察药物的有效期,将三批受试制剂按上市时的包装,置一般药库中,按一定日期取样测定,通常是在0月、3月、6月、9月、12月、18月、24月取样,观察其有关质量指标的变化,如无明显变化的,仍应继续考察,以提供稳定性的详细资料,对不稳定的药物,应通过研究制订贮藏条件及有效期。
5 药效学研究
药效学试验是预测药物疗效的重要手段,应围绕其拟定的临床适应症展开相应的药效学评价,评价方法应遵照目前业界公认的或经验证后的方法、模型,根据研究结果判定药物制剂的作用强弱及主要的作用特点,为临床应用提供科学依据。药物临床前药效学研究试验,应当采用制备工艺稳定、符合临床试验质量标准规定的供试样品。试验设计应符合随机、对照、重复的原则。由于中药制剂本身情况比较复杂,在进行研究时,应遵循“具体问题具体分析”的原则。
6 安全性研究
6.1 急性毒性试验 急性毒性试验是研究动物一次或24 h内多次接受一定剂量的受试物,在一定时间内出现的毒性反应。其主要目的是了解受试药急性毒性的强度、为药效学研究提供剂量参考、为临床毒副反应的监测及解救提供参考依据、为长期毒性、蓄积毒性试验选择剂量提供依据以及用于质量控制等。急性毒性试验处在药物毒理研究的早期阶段,对阐明药物的毒性作用和了解其毒性靶器官具有重要意义。
根据所观察到的各种反应出现的时间、严重程度、持续时间等,分析各种反应在不同剂量时的发生率、严重程度。根据观察结果归纳分析,考察每种反应的剂量-反应及时间-反应关系。急性毒性试验一般测定LD 50或最大给药量。判断出现的各种反应可能涉及的组织、器官或系统等。根据大体解剖中肉眼可见的病变和组织病理学检查的结果,初步判断可能的毒性靶器官。如组织病理学检查发现有异常变化,应附有相应的组织病理学照片。根据不同剂量组各种毒性反应及发生率、动物死亡情况等,确定动物对受试物的毒性反应的性质与强度。根据急性毒性试验结果,提示在其他安全性试验、临床试验、质量控制方面应注意的问题;同时,结合其他安全性试验、有效性试验及质量可控性试验结果权衡利弊,分析受试药物的安全性。
6.2 长期毒性试验 长期毒性试验是非临床安全性评价的重要内容,描述动物重复接受受试药物后的毒性特征。其主要目的是预测受试物可能引起的临床不良反应,包括不良反应的性质、程度、剂量-反应和时间-反应关系、可逆性等;预测临床试验的起始剂量和重复用药的安全剂量范围;提示临床试验中需重点监测的指标;推测受试物重复给药的临床毒性靶器官或靶组织;为临床试验中的解毒或解救措施提供参考信息。根据长期毒性试验结果,分析讨论需在临床、质量可控性研究中注意的问题。
7 小结
医疗机构中药制剂以其处方有特色、临床疗效确切、可满足疾病的个性化治疗需要,弥补市售中成药的不足而被广大临床医师及患者所接受。医疗机构中药制剂是经过长期临床实践检验的成果,体现各医疗机构中医药特色优势,也是中药新药创制的源泉[5]。
提取工艺路线的确定主要是根据处方药物有效成分或有效部位的理化性质和药理作用,制剂的剂型,结合临床要求,生产可行性和生产设备等多方面的因素,设计合理的工艺路线。在传统中医药理论指导下进行医疗机构中药制剂的开发,继承中药临床宝贵经验,借助现代科技手段,使相关的研究成果与中医药的创新结合,有利于加速中医药现代化进程。
通常医疗机构中药制剂均利用传统工艺配制,配制过程中保留原组方中治疗疾病的物质基础,且在本医疗机构使用5年以上,处方组成不含有法定标准中标识有毒性或现代毒理学证明有致癌、致畸、肝、肾毒性等具有毒性作用的药材,无十八反、十九畏配伍禁忌,药味用量不超过法定药品标准规定的,如果该制剂不用于儿童、孕妇等特殊人群可以减免药效学和安全性研究。
孙冬梅教授从事医疗机构制剂研发多年,有丰富的研究经验,特别是医疗机构中药制剂质量标准研究方面,取得了显著成绩,为广东省多家医院提高医院制剂质量标准,保证了临床用药的安全、有效,为广东省医疗机构中药制剂研究提供了范例。
[1]夏杰,尹蔚萍,张文荫.发展中医医院中药制剂的思考[J].中医药管理杂志,2014,22(9):1523-1527.
[2]王红莉,潘五九,王伟明.中药新药的处方筛选研究进展[J].黑龙江中医药,2015,5:75-76.
[3]李钧.药品GM实施与认证[M].北京:中国医药科技出版社,2000.
[4]梁小银,陈少旭,吴垠.中药制剂质量控制研究的发展趋势[J].中国药房,2014,25(3):280-283.
[5]梁建文.浅谈中药特色技术传承项目在医院中药制剂的应用[J].教育教学论坛,2017,(49):213-214.
