癫痫性脑病的遗传学研究进展
2018-06-19孟淑淑褚旭孔庆霞
孟淑淑 褚旭 孔庆霞○☆
癫痫性脑病是一组疾病的总称,其临床类型多种多样,诊断明确较为困难,导致癫痫性脑病在诊疗时给临床医生增加了负担。目前明确的癫痫性脑病有10种,包括:早期肌阵挛脑病(early myoclonic encephalopathy,EME)、大田原综合征 (ohtahara syndrome,OS)、West综合征 (West syndrome,WS)、Dravet综合征 (Dravet syndrom,DS)、Lennox—Gastaut综合征(Lennox gastaut Sydrome,LGS)、获得性癫痫性失语(landau kleffner syndrome,LKS)、癫痫伴慢波睡眠期持续棘慢波 (continuous spike-wave discharges during slow wave sleep,CSWS)、婴儿癫痫伴游走性局灶性发作(epilepsy of infancy with migrating focal seizures,EIMFS)、肌阵挛-站立不能性癫痫 (epilepsy with myoclonic astatic seizures,MAE)和腊斯默森综合征(Rasmussen syndrome,RS)[1]近年来,随着基因检测技术的进步,部分基因突变所致的癫痫性脑病已在临床中得到证实。广泛使用的二代基因测序技术,即全外显子测序和全基因组测序,很大程度上增加了癫痫性脑病突变基因的检出率,本文对癫痫性脑病的遗传因素做出总结,为癫痫性脑病的诊断提供有力证据。
1 早期肌阵挛脑病
197 8 年由 AICARDI和GOUTIERES[2]首次报道,患儿主要表现为肌阵挛,即四肢远端、面部肌肉的痉挛,尤其是游走性的肌阵挛及频繁的部分性发作。脑电图主要表现为睡眠中的爆发抑制,并伴有严重的智力和运动发育障碍。
关于其遗传学病因,其部分病例可能属于常染色体隐性遗传,如非酮症高甘氨酸血症等[3-4],发现的基因有AMT c.793C> T(Arg265CYS)。 在 KOJIMA 等[5]报道的 EME 中存在 KCNQ2 c.601C>T(Arg201Cys)基因突变,导致电压门控钾通道产生M电流,其特征为慢激活或去激活电导,M电流的较小改变,就可诱发癫痫发作。在COHEN等[6]的报道中, 发现了 SLC25A22 c.706G> T (Gly236Trp)、c.617C> T(Pro206Leu)基因发生了错义突变,SLC25A22编码的蛋白在脑的脑桥小脑角等处高度表达,在星形胶质细胞中最为丰富,控制线粒体内膜谷氨酸摄取并催化谷氨酸转运蛋白转运。SLC25A22基因功能丧失的个体,导致细胞外谷氨酸的功能失调,导致神经元功能异常或坏死。LIESBETH等[7]报道的神经调节蛋白1受体ErbB4发生的碱基异位t(2;6)(q34;p25.3)并指出 ErbB4或神经调节蛋白 1受体功能的丧失扰乱了皮层发育过程中中间神经元的迁移并改变GABA能中间神经元的数量,故解释了患者皮质GABA能中间神经元数量或功能异常的原因。
2 大田原综合征
由日本学者OHTAHARA[8]于1976年首次报道。主要表现为频繁且不易控制的强直性痉挛发作。清醒及睡眠脑电图均呈爆发抑制,伴有严重的精神、智力及运动发育障碍。目前有较多报道基因突变所导致的OS(见表1),多因为离子通道异常或蛋白结构改变所致。其中SCN2A基因突变[9-11]可引起一系列的临床病症,如自闭症和各种癫痫等。SCN2A编码神经元钠离子通道亚基,其中α亚基负责通道的开放、离子选择和通道的快速灭活,其突变的后果为诱发癫痫。KODERA等[12]报道了STXBP1基因基因突可致OS。 目前发现的错误位点为:STXBP1 c.247-2A>G、c.902+1G>A、c.246+1G>A、c.1056del Asp353ThrfsX3,多为剪接异常及移码突变。STXBP1编码的蛋白质是突触融合蛋白,并且能对NSF附着蛋白受体的形成、钙通道感受器的敏感性和突触囊泡与突触前膜的融合有调节作用,并且控制突触间神经递质的释放,导致惊厥发作。ARX基因定位于Xp22.13,其编码蛋白在脑中间神经元增生与分化中发挥重要作用。其突变可导致不同的临床表型,包括OS、智力低下、婴儿痉挛等[13]。此外,CDKL5基因定位于染色体Xp22.13,编码具有激酶活性的磷酸化蛋白,可以调节其激酶活性和核酸定位,推测CDKL5基因在神经可塑性中起重要作用。
3 West综合征
于1841年WEST首先详细描述,是婴儿痉挛症(infantile spasms,IS)的一个亚型[14]。成串痉挛发作是本病独有的特点,每次痉挛持续时间为1~2 s,一次抽搐后间隔1~2 s又发作第2次,可连续十余次甚至上百次。
目前研究表明,与神经发育有关的基因突变常常会导致此病的发生(见表2)。并且许多基因的突变与其他类型的癫痫性脑病有一致性。其中具有特异性的基因有NMD1、MAGI2、TSC2。NMD1 基因突变,导致发动蛋白-1 与GTP的结合或者抑制GTP水解活性都能使内吞和突触囊泡循环过程出现异常,导致癫痫的发生。

表1 OS相关基因突变
4 Dravet综合征
由DRAVET[18]于1978年首次报道,是一种少见遗传因素导致的癫痫性脑病。DS患儿早期脑电图多正常,1岁以后出现,背景活动异常,以慢波增多为主,随后出现全导棘慢波或多棘慢波。对于其遗传因素,最突出的致病基因有SCN1A和SCN2A(见表3)。其编码非电压门控性钠离子通道α亚单位。 SCN1A的位点主要为:c.5732T>G(lle1911ser)、c.719T>C(leu240pro)、c.1262A>G(Val 421Ala)、c.2378G >A (Thr793Met)、c.311C >T (Ala104Val)、c.2948delT(Val983Alafs*2)等。SCN1B主要编码非电压门控性钠离子通道β亚单位,通过改变电压敏感性、调节失活过程等调控α亚单位的功能。因为通道的异常开放,导致了癫痫神经兴奋性网络的形成[17-21-22]。GABRB3可介导中枢神经系统内的抑制性信号传导,基因发生突变时,可导致兴奋性氨基酸相对过多,引起癫痫发作[18]。编码GABAA受体γ2亚基的GABRG2突变可导致遗传性癫痫伴热性惊厥和Dravet综合征[19]。
5 Lennox—Gastaut综合征
LGS是严重的年龄依赖性癫痫性脑病,其临床表现复杂多样,包括思维缓慢、言语不清、智力倒退、认知障碍、行为异常等。脑电图通常显示背景活动异常,发作间期有3HZ的慢棘慢波,睡眠期可有快节律爆发出现。
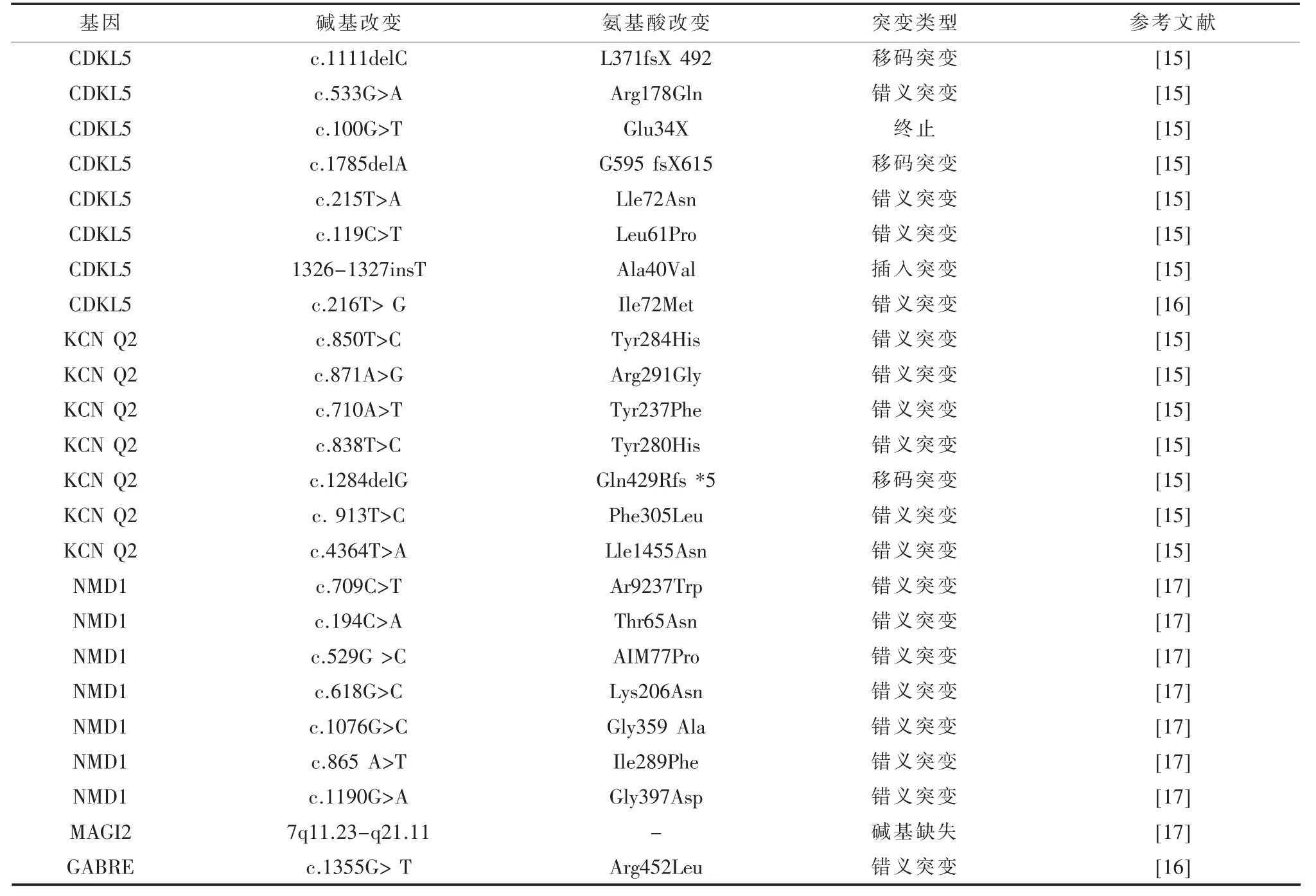
表2 West综合征相关基因突变

表3 Dravet综合征相关基因突变
LUND等[26]报道了LGS患者中染色体的杂合缺失22q13和2q23.1,影响SHANK3。其在兴奋性突触的形成和成熟过程中起到重要作用。编码多种蛋白,如NMDA受体、代谢型谷氨酸受体,其过表达可引起痫性发作。DELMIR0等[27]报道了MTND1基因,作用于成纤维细胞线粒体呼吸链复合体1中,并且导致其功能的缺陷。CAROLINE等[28]报道了CHD2可通过结合RE1沉默转录因子的启动子直接调控REST的转录,从而调控神经前体细胞的增殖与分化。关于NMD1、GRIN2A、GABRB3导致的癫痫发作上文已经描述,目前发现的位点有:DNM1c.127G>A (Gly43Ser)、c.709C>T(Arg237Trp)、DNM1 c.529G>C (Ala177Pro)DNM1c.1076G>C(Gly359Ala)等[31]。 此外,GPR56 c.1693 C>T(Arg565Trp)、c.235 C>T(Arg79STOP)、c.97 C>G(Arg33Phe)基因突变致病可能通过影响少突胶质前体细胞分化成熟(见表4)。
6 获得性癫痫性失语
LANDAU[32]和KLEFFNER于1957年首次报道,临床特征表现为急性或亚急性听觉和认知能力丧失并伴有阵发性脑电图异常,其中以听觉失认最为多见。脑电图异常是LKS最明确的诊断指标,表现为背景节律正常或轻度异常,一侧或双侧颞、枕区出现棘波、尖波,睡眠期全导泛化。对于遗传学方面,GRIN2A基因突变导致癫痫发作,目前发现以下位点:GRIN2A c.2041C>T(Arg681*)、c.1007+1G>A IVS4、c.692G>A (Cys231Tyr)、c.2191G>A (Asp731Asn)、c.2T>C(Met1Thr)、c.1007+1G>A(Phe139Ilefs*15)[33]。CONROY J 等还确立了其他一些候选基因,即 RELN,BSN,EPHB2和NID2等,最明确的是 EPHB2 c.464G>A(Arg155His)。 其编码的受体蛋白是酪氨酸激酶家族中的一员,在中枢神经系统中主要调节神经嵴的迁移、轴突导向和突触发生。
7 癫痫伴慢波睡眠期持续棘慢波
PATRY等[34]1971年首次报道,并认为是儿童期发生的严重影响认知功能的癫痫综合征,表现为癫痫发作、全面性脑功能减退及脑电图慢波睡眠期持续性癫痫性电活动(ESES),发病率占儿童期癫痫的0.2%~0.5%。颅脑MRI大多无明显异常。关于GRIN2A、KCNQ2导致的癫痫发作上文已阐述,现将具体的基因突变阐述见表5。

表4 Lennox-Gastaut综合征相关基因突变

表5 癫痫伴慢波睡眠持续棘慢波相关基因突变
8 婴儿癫痫伴游走性局灶性发作
由意大利学者COPPOLA[38]于1995年首次报道,其临床特点为频繁的、游走性的、多种类型的局灶性发作;脑电图发作期表现为多灶性起源的局灶性发作;智力、运动发育落后或倒退。
近年来,发现了越来越多的EIMFS致病基因(见表6)。SAITO 等[39]报道了 SLC12A5 c.7C>T(Arg3Cys)、c.1196C>T(Ser399Leu)c.2639G>T(Arg880Leu)位点出现错义突变导致的癫痫发作。SLC12A5编码KCC2通道蛋白,在神经元中特异性表达,其中,KCC2结构改变,可能使神经元去极化,并阻碍抑制性突触后电位的产生,进而导致突触抑制受损和过度的神经元兴奋性。同时也描述了其GPR78基因突变也可能导致癫痫发作,余其他基因突变导致的癫痫诱发机制上文已描述。
9 肌阵挛-站立不能性癫痫和腊斯默森综合征
MAE及RS在临床工作中尚不多见,近年来,TANG S等报道了MAE患者中存在SCN1A、SCN1B和 GABRG2突变,考虑是导致MAE发病的原因。VLASKAMP等描述了在染色体16p11.2上的1.2-Mb缺失的STX1B基因导致了MAE的发生。但对于RS目前尚无明确遗传学资料证实与基因突变有关。
综上所述,随着基因测序技术的进步,癫痫性脑病的病因在分子水平上有了更深层次的认识,但是各个类型的癫痫性脑病常常有相同的突变基因及相似的临床表现,仍给我们诊断带来了困难,因此诊断癫痫性脑病,仍要根据患者发病年龄、临床症状、脑电图、颅脑MRI及治疗效果等综合分析。但是我们相信在不久的将来,遗传学的进步与发展,会给予我们带来福音。
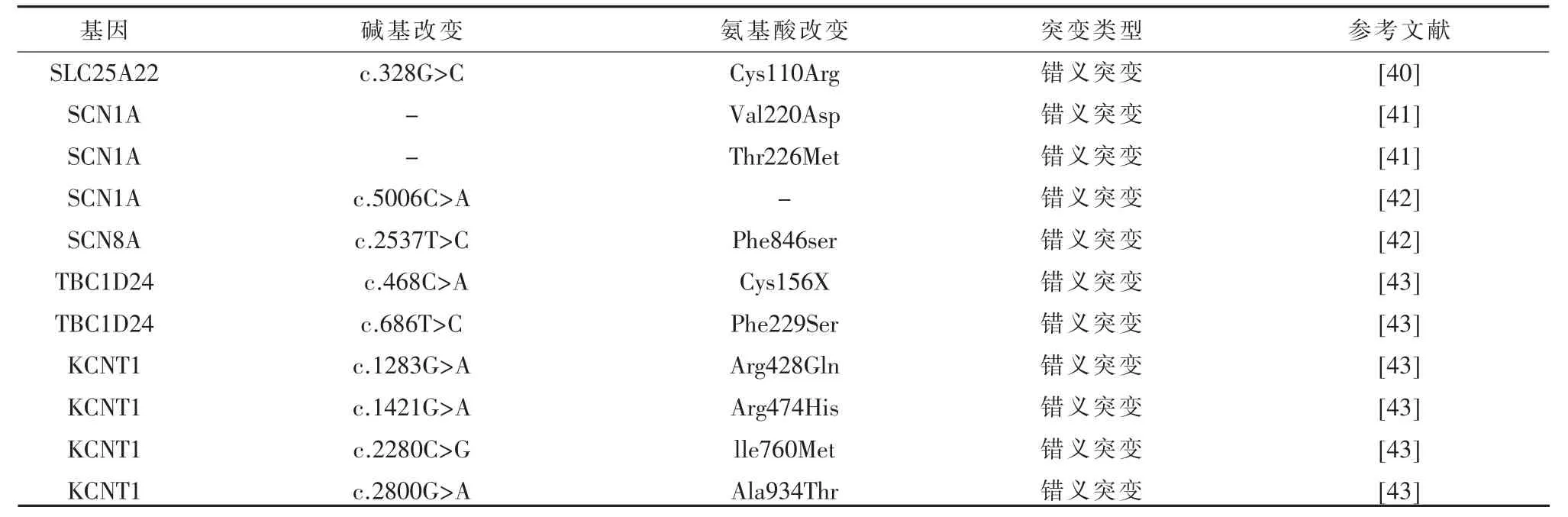
表6 婴儿癫痫伴游走性局性发作相关基因突变
[1]邓劼,张月华,刘晓燕,等.发作和癫痫分类框架相关术语和概念修订-国际抗癫痫联盟分类和术语委员会报告[J].中国实用儿科杂志,2011,26(07):505-511.
[2]AICARDI J,GOUTIERES F.Neonatal myoclonic encephalopathy(author's transl)[J].Electroencephalogr Neurophysiol Clin,1978,8(1):99-101.
[3]ROSSI S,DANIELE I,BASTRENTA P,et al.Early myoclonic encephalopathy and nonketotic hyperglycinemia [J].Pediatr Neurol,2009,41(5):371-374.
[4]VINCENZO B,MARIO B,SALVATORE B,et al.A novel AMT gene mutation in a newborn with nonketotic hyperglycinemia and early myoclonic encephalopathy[J].Eur J Paediatr Neurol,2016,20(1):192-195.
[5]KOJIMA K,SHIRAI K,KOBAYASHI M,et al.A patient with early myoclonic encephalopathy(EME)with a de novo KCNQ2 mutation[J].Brain Dev,2018,40(1):69-73.
[6]COHEN R,BASEL-VANAGAITE L,GOLDBERG-STERN H,et al.Two siblings with early infantile myoclonic encephalopathy due to mutation in the gene encoding mitochondrial glutamate/H+symporter SLC25A22[J].Eur J Paediatr Neurol,2014,18(6):801-805.
[7]LIESBETH B,BERTEN C,JORIS RV,et al.Early myoclonic encephalopathy caused by a disruption of the neuregulin-1 receptor ErbB4[J].Eur J Hum Genet,2009,17(3):378-382.
[8]OHTAHARA S,OHTSUKA Y,OKA E.Epilepticencephalopathies in early infancy[J].indian J pediatr,1997,64(05):603-612.
[9]ZEREM A,LEV D,BLUMKIN L,et al.Paternal germline mosaicism of a SCN2A mutation results in Ohtahara syndrome in half siblings[J].Eur J Paediatr Neurol,2014,18(5):567-571.
[10]NAKAMURA,KATO M,OSAKA H,et al.Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome[J].Neurology,2013,81(11):992-998.
[11]HOWELL KB,MCMAHON JM,CARVILL GL,et al.SCN2A encephalopathy:A major cause of epilepsy of infancy with migrating focal seizures[J].Neurology,2015,85(11):958-966.
[12]KODERA H,KATO M,NORD AS,et al.Targeted capture and sequencing for detection of mutations causing early onset epileptic encephalopathy[J].Epilepsia,2013,54(7):1262-1269.
[13]PAVONE P,SPALICE A,POLIZZI A,et al.Ohtahara syndrome with emphasis on recent genetic discovery[J].Brain Dev,2012,34(6):459-468.
[14]PELLOCK JM,HRACHOVY R,SHINNAR S,et al.Infantile spasms:a US consensus report[J].Epilepsia,2010,51(10):2175-2189.
[15]ZHANG Q,LI J,ZHAO Y,et al.Gene mutation analysis of 175 Chinese patients with early-onset epileptic encephalopathy[J].Clin Genet,2017,91(5):717-724.
[16]WANG Y,DU X,BIN R,et al.Genetic Variants Identified from Epilepsy of Unknown Etiology in Chinese Children by Targeted Exome Sequencing[J].Sci Rep,2017,(7):40319.
[17]邓小鹿,尹飞,张慈柳,等.Dynamin-1基因新生突变导致婴儿痉挛症一例并文献复习[J].中华儿科杂志,2016,54(11):856-859.
[18]Li BM,Liu XR,Yi YH,et al.Autism in Dravet syndrome:prevalence,features,and relationship to the clinical characteristic of epilepsy and mental retardation[J].Epilepsy Behav,2011,21:291-295.
[19]杨李,吴德,唐久来.Dravet综合征3例临床及分子遗传学研究[J].中国儿童保健杂志,2016,24(10):1037-1040.
[20]LE SV,LE PHT,LE TKV,et al.A mutation in GABRB3 associated with Dravet syndrome[J].Am J Med Genet A,2017,173,(8):2126-2131.
[21]ISHII A,KANAUMI T,SOHDA M,MISUMI Y,et al.Association of nonsense mutation in GABRG2 with abnormal trafficking of GABAA receptors in severe epilepsy[J].Epilepsy Res,2014,108(3):420-432.
[22]INGO HA,C AHMAD N,ABOU T,et al.Understanding Genotypes and Phenotypes in Epileptic Encephalopathies[J].Mol Syndromol,2016(7):172-181.
[23]MUKHERJEE D,MUKHERJEE S,NIYOGI P,et al.Dravet syndrome with SCN1B gene mutation:A rare entity[J].Neurol India,2017,65(4):801-803.
[24]LOSSIN C,SHI X,ROGAWSKI MA,et al.Compromised function in the Na(v)1.2 Dravet syndrome mutation R1312T[J].Neurobiol Dis,2012,47(3):378-384.
[25]HARKIN LA,BOWSER DN,DIBBENS LM,et al.Truncation of the GABA(A)-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus[J].Am J Hum Genet,2002,70(2):530-536.
[26]TRIVISANO M,STRIANO P,SARTORELLI J,et al.CHD2 mutations are a rare cause of generalized epilepsy with myoclonic-atonic seizures[J].Epilepsy Behav,2015,51(3):53-56.
[27]邓小鹿,何芳,吴丽文,等.CHD2基因突变导致Dravet综合征1例病例报告[J].中国循证儿科杂志,2016,12(11):473-474.
[28]LUND C,BRODTKORB E,RQSBY O,et al.Copy number variants in adult patients with Lennox-Gastaut syndrome features[J].Epilepsy Res,2013,105(1-2):110-117.
[29]DELMIRO A,RIVERA H,GARCÍA-SILVA MT,et al.Wholeexome sequencing identifies a variant of the mitochondrial MTND1 gene associated with epileptic encephalopathy:west syndrome evolveng to Lennox-Gastaut syndrome[J].Hum Mutat,2013,34(12):1623-1627.
[30]BERNARDO P,GALLETTA D,IASEVOLI F,et al.CHD2 mutations:Only epilepsy?Description of cognitive and behavioral profile in a case with a new mutation[J].Seizure,2017,51(3):186-189.
[31]NAKASHIMA M,KOUGA T,LOURENÇO CM,et al.De novo DNM1 mutations in two cases of epileptic encephalopathy[J].Epilepsia,2016,57(1):18-23.
[32]LANDAU WM,KLEFFNER FR.Syndrome of acquired aphasia with convulsive disorder in children[J].Neurology.1957,7(8):523-530.
[33]LEMKE JR,LAL D,REINTHALER EM,et al.Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes[J].Nat Genet,2013,45(9):1067-1072.
[34]PATRY G,LYAGOUBI S,TASSINARI CA,et al.Subclinical electrical status epilepticusinduced by sleep in children:a clinical and electroencephalographic study of six cases[J].Arch Neurol,1971,24:242-252.
[35]LEMKE JR,LAL D,REINTHALER EM,et al.Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes[J].Nat Genet,2013,45(9):1067-1072.
[36]CARVILL GL,REGAN BM,YENDLE SC,et al.GRIN2A mutations cause epilepsy-aphasia spectrum disorders[J].Nat Genet,2013,45(9):1073-1076.
[37]TURNER SJ,MORGAN AT,PEREZ ER,et al.New genes for focal epilepsies with speech and language disorders[J].Curr Neurol Neurosci Rep,2015,15(6):35.
[38]COPPOLA G,PLOUIN P,CHIRON C,et al.Migrating partial seizures in infancy:a malignant disorder with developmental arrest[J].Epilepsia,1995,36:1017-1024
[39]SAITO T,ISHII A,SUGAI K,et al.A de novo missense mutation in SLC12A5 found in a compound heterozygote patient with epilepsy of infancy with migrating focal seizures[J].Clin Genet,2017,92(6):654-658.
[40]PODURIA,HEINZEN EL,CHITSAZZADEH V,etal.SLC25A22 is a novel gene for migrating partial seizures in infancy[J].Ann Neurol,2013,74(6):873-882.
[41]FREILICH ER,JONES JM,GAILLARD WD,et al.Novel SCN1A mutation in a proband with malignant migrating partial seizures of infancy[J].Arch Neurol,2011,68(5):665-671.
[42]NIEH SE,SHERR EH,et al.Epileptic encephalopathies:new genes and new pathways[J].Neurotherapeutics,2014,11(4):796-806.
[43]DE FILIPPO MR,RIZZO F,MARCHESE G,et al.Lack of pathogenic mutations in six patients with MMPSI[J].Epilepsy Res,2014,108(2):340-344.
