基于高密度Bin图谱的水稻抽穗期QTL定位
2018-06-15董骥驰陈立凯陈志强
董骥驰 杨 靖 郭 涛 陈立凯 陈志强 王 慧
基于高密度Bin图谱的水稻抽穗期QTL定位
董骥驰**杨 靖**郭 涛 陈立凯 陈志强*王 慧*
华南农业大学 / 国家植物航天育种工程技术研究中心, 广东广州 510642
以粳稻品种02428和籼稻品种玉针香进行杂交, 按单粒传法连续自交10代, 得到包含192个株系的重组自交系(RIL)作图群体。通过对两亲本重测序及RIL群体简化基因组测序, 构建了包含2711个Bin标记的高密度遗传图谱。该图谱各染色体标记数在162~311个之间, 标记间平均物理距离为137.68 kb。将亲本及192个株系分别于4个环境下采用随机区组种植, 并记录抽穗期。使用WinQTL Cartographer 2.5软件的CIM分析方法, 进行抽穗期相关QTL检测及定位。在4个环境下定位到影响抽穗期的QTL共14个, 分布于第1、第2、第3、第7、第8、第9和第10染色体。其中,和能在3个环境中被重复检测到, 表型贡献率分别为5.14%~11.15%和5.35%~16.97%, 分别能缩短抽穗期1.66 d和1.56 d, 具有聚合育种的应用价值。通过物理位置比对, 14个QTL中有11个与前人定位在相同或邻近区域,和尚未见报道。经对详细分析, 在其染色体区间内找到3个与抽穗期相关的注释基因、和, 其中已被克隆。测序分析发现, 这3个基因在两亲本间都存在差异, 可作为候选基因。
水稻; 抽穗期; Bin图谱; QTL定位
水稻抽穗期决定了品种的种植地区与季节适应性[1], 而且与产量、品质和抗逆性关系密切[2], 相关的研究对指导育种实践、品种改良及品种推广均具有重要意义[3]。自Yano等克隆了第一个水稻抽穗期QTL ()以来, 目前至少已有734个与水稻抽穗期相关的QTL被报道(http://www.grammene.org/), 用图位克隆的方法至少克隆了14个抽穗期相关的QTL[4-17]。已有研究表明, 水稻有2个成花素基因()和(), 以及几个抽穗抑制基因(如和), 它们形成的调控网络精密控制着抽穗。至少有2个抽穗期调节通路控制着成花素基因表达, 分别是()通路和()通路。大量研究证实, 大多数抽穗期相关基因通过通路或通路来调节成花素基因的表达,和则不同, 它们独立于和通路, 直接调节成花素基因的表达进而促进抽穗[18]。然而, 已定位和克隆的QTL还并不能完全解释水稻抽穗期自然变异的形成, 从根本上理解水稻抽穗期的遗传机理还需更多的深入研究, 从而更好地帮助适宜生育期品种的选育[19-20]。目前的研究大多基于传统分子标记(如AFLP、RFLP、SSR等), 操作起来耗时耗力, 不能实现高通量操作[21]。用传统分子标记构建的遗传图谱密度低、定位区间过大, 导致QTL贡献率估算偏高、基因克隆困难以及难以开发标记应用于聚合育种。此外, 标记密度低的图谱上存在大量Gap, 在QTL定位中不能提供足够的信息, 甚至一些研究最终陷入多态性分子标记缺乏而难以进一步定位的境地[22]。本研究中的Bin标记分布于整个基因组, 构建的Bin图谱标记密度高、位置精确, 达到精细定位要求, 可直接进行分子育种标记的开发和候选基因的筛选。本研究以籼稻玉针香(YZX)和粳稻02428衍生的RIL为作图群体, 采用GBS技术构建包含2711个Bin标记的高密度遗传图谱, 分别于4个环境对水稻抽穗期进行QTL定位, 以期鉴定一些新的、可稳定遗传的QTL位点作为育种资源, 并为水稻抽穗期调控基因克隆及分子标记辅助育种提供更多依据。
1 材料与方法
1.1 试验材料
用籼稻玉针香(YZX)和粳稻02428杂交, 通过单粒传法, 从F2代开始构建玉针香/02428的重组自交系群体。群体包含192个株系, 基因型鉴定为F6世代, 表型调查为F8~F11世代。
1.2 田间种植与性状考查
2016年早造(E1环境)、2016年晚造(E2环境)、2017年早造(E3环境)和2017年晚造(E4环境)于华南农业大学试验教学基地(23.17°N, 113.37°E)种植192个株系的RIL群体及两亲本。采用随机区组设计, 田间管理(水、肥、病虫害防治等)按当地大田常规栽培要求实施。E1环境, 于2016年3月5日播种育秧, 4月5日移栽。E2环境, 于2016年7月25日播种育秧, 8月8日移栽。E3环境, 于2017年2月28日播种育秧, 3月31日移栽。E4环境, 于2017年7月22日播种育秧, 8月7日移栽。每个株系6行, 每行6株, 行株距为20 cm × 20 cm, 均单本种植。
以单株为单位调查表型, 当单株的第1个稻穗尖露出剑叶叶鞘至少1 cm时, 记为该单株的抽穗日期, 每隔1 d调查一次。剔除异常数据后, 以株系内全部单株从播种到抽穗所经历天数的均值作为该株系的抽穗期表型值。
1.3 DNA提取及高通量测序
采用本实验室自主开发设计的高通量磁珠法制备DNA样品。用Precellys 24研磨仪(4000´, 12 s)研磨液氮冷冻过的叶片至粉末, 迅速加入85℃预热的LB提取液(200 mmol L–1Tris-HCl (pH 7.8), 250 mmol L–1NaCl, 25 mmol L–1ethylenediaminetetraacetic acid (EDTA), 0.5% sodium dodecyl sulfate (SDS), 2% polyvinylpyrrolidone (PVP)-40), 确保叶片粉末都浸泡在提取液中, 迅速盖上橡胶盖并混匀, 置于65℃水浴锅中水浴20~25 min后, 置–20℃冰箱放置20 min。将样品转移至96孔深孔板, 每孔加入540 µL异丙醇和2.5 µL磁珠液(100 mg mL–1, 洛阳惠尔纳米科技有限公司), 用自动核酸纯化仪KingFisher Flex进行DNA分离纯化。DNA最终溶于150 μL洗脱液(10 mmol L–1Tris-HCl, 1 mmol L–1EDTA (pH 8.3), 100 μg mL–1RNase A), 于–80℃保存备用。以琼脂糖凝胶电泳检测基因组DNA的质量, Nanodrop ND-1000微量核酸蛋白检测仪检测DNA的浓度与纯度[23]。
采用WGS (Whole Genome Sequencing)和GBS (Genotyping-By-Sequencing)技术分别对两亲本及其衍生的RIL群体测序。水稻基因组经电子酶切评估, 选择酶切片段大小适宜, 酶切位点在基因组上分布均匀, 且能有效降低酶切片段中基因组重复序列所占比例的限制性内切酶。基因组加上带有barcode的接头后, 对每个样品进行扩增, 随后混合选择需要的片段构建GBS文库。利用Illumina HiSeq2500测序平台, 进行双末端(Paired-End)测序。测序后的数据分析流程如下: (1)数据质量评估, 去除接头、污染序列及低质量reads; (2)统计酶捕获的Reads数量; (3)参考基因组(http://plants.ensembl.org/Oryza_ sativa/)比对: 利用BWA软件(http://bio-bwa.sourceforge. net/)将测得基因组与参考基因组比对, 统计比对率、覆盖深度、基因组覆盖率等; (4)群体SNP检测; (5)遗传标记开发。基于亲本SNP检测结果, 开发子代标记, 并统计子代测序深度、覆盖度等, 评估测序质量。与亲本玉针香相同的基因型记为“0”, 与亲本02428相同的基因型记为“2”, 部分杂合基因型记为“1”。(6)遗传标记过滤。检查异常碱基, 检查基因型缺失覆盖率并过滤, 检查基因型频率, 过滤偏分离标记。
1.4 遗传重组鉴定和连锁图谱构建
采用ScmapV5软件构建遗传图谱。构建原理是: (1)通过“滑动窗口”法(sliding window approach)寻找交换点; (2)将所有样本中同一段序列上2个交换点之间紧密连锁不发生重组的若干个SNP位点看做一个整体区块, 即“Bin标记”, 以Bin的起点来确定其所在物理位置; (3)根据基因型, 用最大似然函数计算重组率; (4)用Kosambi作图函数将重组率转换为遗传距离; (5)利用两点检验法检验两个标记间是否连锁。(6)通过最短距离法进行聚类分析, 把两两连锁的标记聚为一个类, 得到连锁群; (7)基于三点测验法, 对连锁群内的标记进行排序。
1.5 数据的统计分析
在Microsoft Excel 2010中完成表型数据处理和作图。采用BWA软件的默认设置进行基因组比对及相关测序数据统计分析。采用WinQTL Cartographer 2.5软件定位水稻抽穗期QTL, 以LOD=2.5为阈值检测QTL; 应用CIM算法, 扫描区间为1.0 cM, 遗传背景控制选择标准模型(模型6)。QTL定位结果以LOD的峰值作为该QTL的LOD值, 以LOD峰值位置的Bin标记来估计QTL效应, 以LOD值下降1.0的区域作为QTL的置信区间, 遵循McCouch等[24]的原则命名QTL。应用SPSS 19.0软件进行方差分析及计算。应用Origin 8.0进行频次分布图的绘制。
2 结果与分析
2.1 遗传连锁图谱构建
亲本玉针香和02428重测序Q20的比例分别为93.72%和94.22%, Q30比例分别为86.44%和87.47%。两亲本测序Reads与Nipponbare (ssp)参考基因组的比对率分别为96.29%和97.97%, 平均深度分别达到20.26×和26.21×, 1×以上覆盖度为90.60%和96.36%, 4×以上覆盖度为85.41%和94.53%。两亲本中共发现1 534 036个多态性SNP, 筛选后剩余aa×bb型1 334 454个。
利用GBS技术对192个RIL株系进行测序, 测序深度平均值为11.76×, 4×以上覆盖度为7%。分别提取192个子代在上述的1 334 454个亲本多态性标记位点的基因型, 经过分析和筛选最终共获得2711个均匀分布于12条染色体的Bin标记。将Bin标记锚定于连锁群后, 各染色体上标记数量为162~311个, 各染色体的平均遗传距离为195.3 cM, 两标记间遗传距离平均值为0.86 cM, 两标记间物理距离平均值为137.36 kb, 标记整体的分布达到精细作图的要求(图1)。
2.2 抽穗期表型特征
方差分析表明两亲本抽穗期天数的均值存在极显著差异, 4个环境下玉针香的抽穗期均显著长于02428 (表1)。
表2表明, 在E1环境中, RIL群体抽穗期的平均值为95.65 d, 抽穗期最短的株系为81.33 d, 抽穗期最长的株系为117.83 d; 在E2环境中, RIL群体抽穗期的平均值为69.15 d, 抽穗期最短的株系为53.13 d, 抽穗期最长的株系为85.4 d; 在E3环境中, RIL群体抽穗期的平均值为95.29 d, 抽穗期最短的株系为81 d, 抽穗期最长的株系为110.3 d; 在E4环境中, RIL群体抽穗期的平均值为70.54 d, 抽穗期最短的株系为61.3 d, 抽穗期最长的株系为87.5 d; 四个环境中RIL群体的抽穗期峰度和偏度绝对值都小于1, 总体呈正态分布(图2), 表现为数量性状遗传模式。
2.3 抽穗期QTL定位
在4个环境中共检测到14个抽穗期QTL(表3和图1)。其中, E1环境中检测到9个, 分布于第2、第3、第7、第8和第10染色体, LOD值介于2.9~5.6之间, 贡献率介于4.7%~9.5%之间; E2环境中检测到5个, 分布于第2、第7和第10染色体, LOD值介于3.0~5.0之间, 贡献率介于5.2%~9.5%之间; E3环境中检测到3个, 分布于第1、第2和第8染色体, LOD值介于2.5~5.9之间, 贡献率介于4.5%~11.2%之间; E4环境中检测到5个, 分布于第3、第9和第10染色体, LOD值介于2.8~9.6之间, 贡献率介于4.6%~17.0%之间。其中, 有2个QTL在3个环境中均检测到。
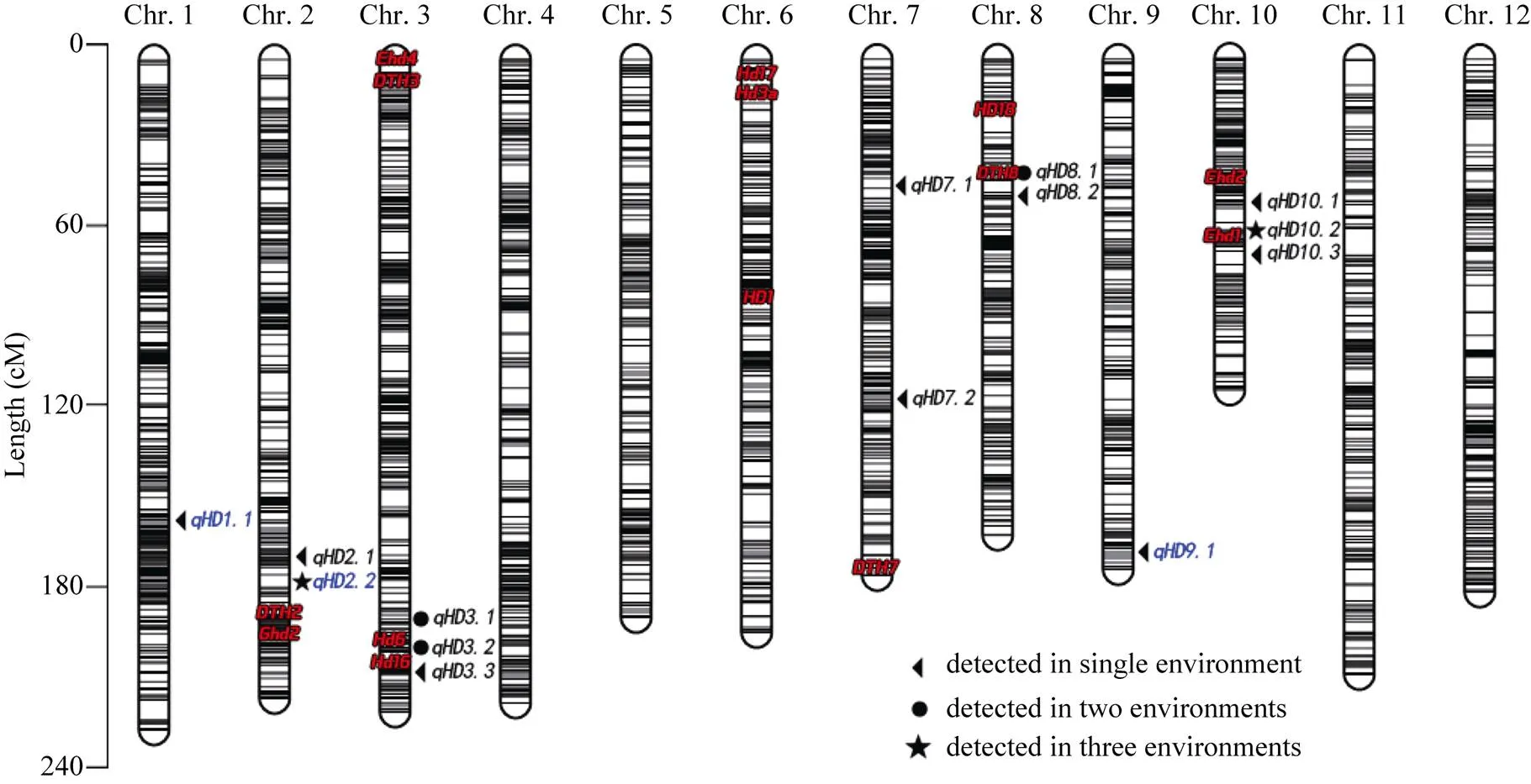
图1 遗传标记在染色体上的分布及QTL位置
每条染色体上的黑色线条代表Bin标记所在位置; 红色字体为已克隆QTL; 蓝色字体为新发现的QTL。
Black lines represent the positions of Bin markers on each linkage group; fonts in red are cloned QTLs; fonts in blue are novel QTLs.
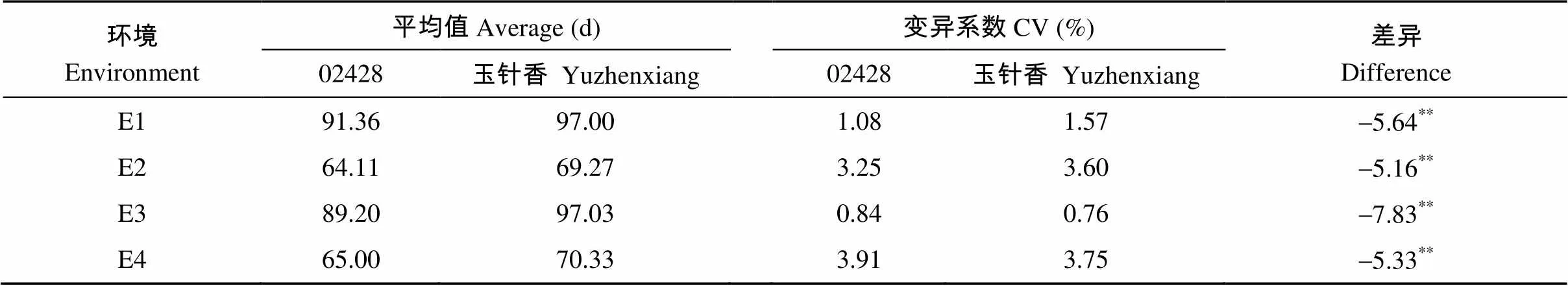
表1 02428与玉针香抽穗期天数比较
**表示差异在0.01水平达到极显著。
**indicates significant difference at the 0.01 probability level.
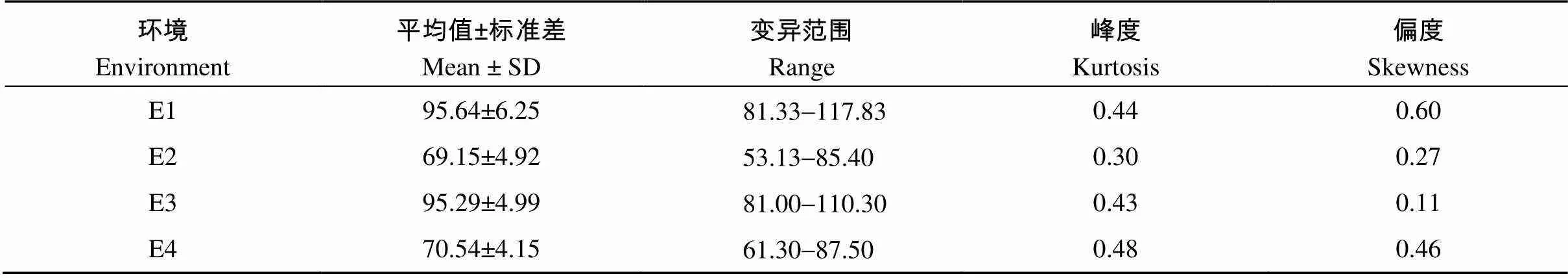
表2 RIL群体抽穗期性状表现
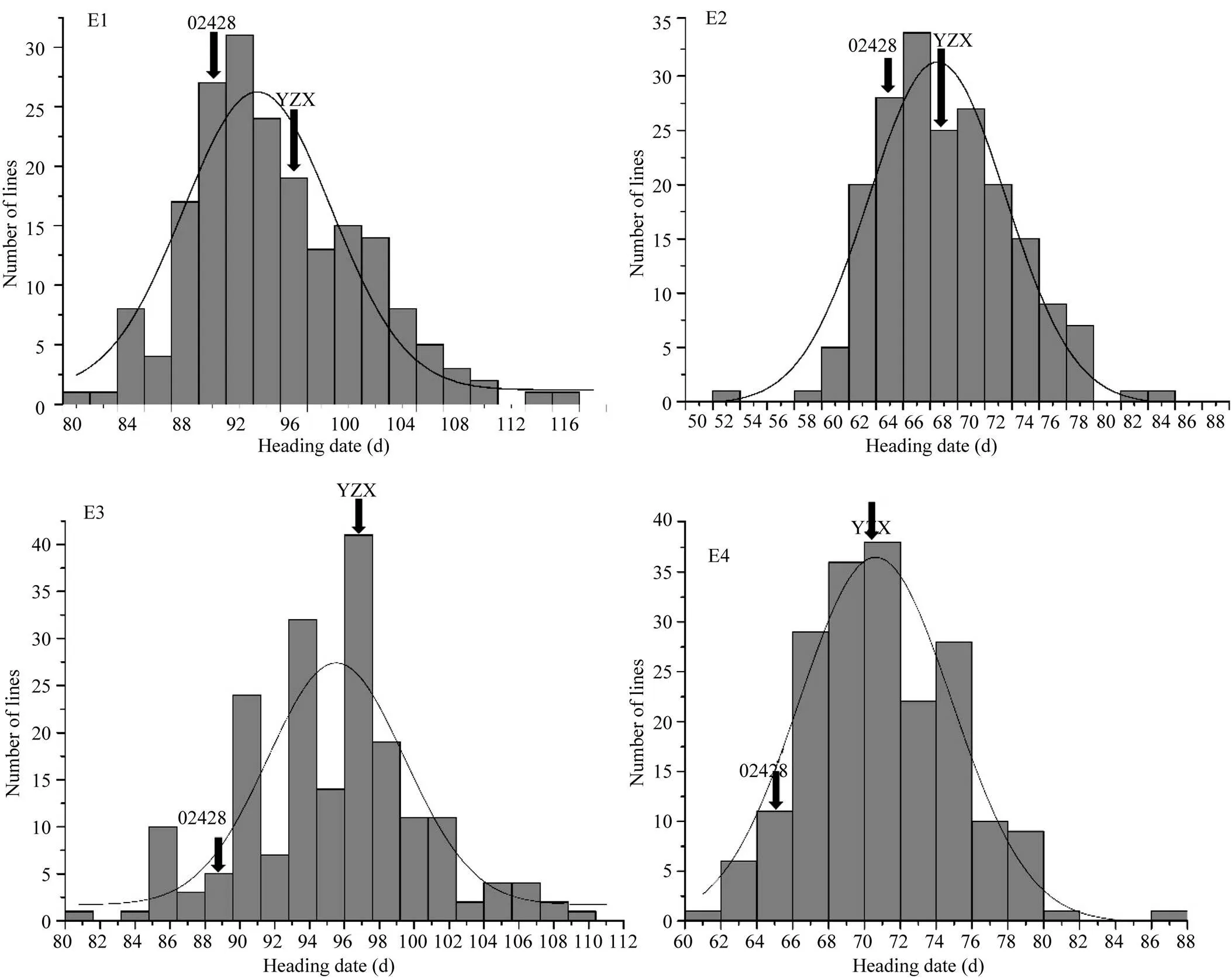
图2 玉针香/02428重组自交系及亲本的抽穗期表型分布特征
在第1染色体上, 仅在E3环境中被检测到, 位于160.17 cM, 与mk186连锁。其LOD值为2.5, 置信区间为25.95~26.05 Mb, 表型贡献率为4.5%, 来自于02428的等位基因可缩短生育期1.3 d。
位于第2染色体, 仅在E2环境中被检测到, 在173.61 cM LOD达到峰值3.2, 与mk484紧密连锁, 置信区间为17.60~28.35 Mb, 能解释5.8%的表型变异, 来自于02428的等位基因可缩短抽穗期1.3 d。同一染色体上的在3个环境中均被检测到, 表现出较强的稳定性。在E1环境检测到的位置是178.21 cM, LOD值为3.1, 置信区间为28.25~28.65 Mb, 可解释5.1%表型变异, 来自于02428的等位基因可缩短抽穗期1.5 d; 在E2环境检测到的位置是179.71 cM, LOD值为5.0, 置信区间为28.45~28.65 Mb, 可解释9.5%表型变异, 来自于02428的等位基因可缩短抽穗期1.6 d。在E3环境检测到的位置是175.19 cM, LOD值为5.9, 置信区间为28.15~28.65 Mb, 可解释11.2%的表型变异, 来自于02428的等位基因可缩短抽穗期1.9 d。
第3染色体上共有3个抽穗期QTL, 分别为和。在E1环境中被检测到, 位于194.21 cM处, LOD值为2.9, 置信区间为30.05~30.65 Mb, 可解释4.7%表型变异, 来自于02428的等位基因可缩短抽穗期1.4 d; 在E4环境中被检测到, 位于197.3 cM处, LOD值为3.4, 置信区间为30.45~31.05 Mb, 可解释5.6%表型变异, 来自于02428的等位基因可缩短抽穗期1.0 d。在E1环境被检测到, 位于205.11 cM处, LOD值为3.2, 置信区间为31.85~32.45 Mb, 可解释5.25%表型变异, 来自于02428的等位基因可缩短抽穗期1.5 d; 在E4环境被检测到, 位于205.11 cM处, LOD值为3.0, 置信区间为31.85~32.45 Mb, 可解释4.85%表型变异, 来自于02428的等位基因可缩短抽穗期1.0 d。仅在E1环境中被检测到, 位于211.21 cM处, LOD值为3.2, 置信区间为33.10~33.55 Mb, 可解释5.3%表型变异, 来自于02428的等位基因可缩短抽穗期1.5 d。
第7染色体上共有2个抽穗期QTL。只在E1环境中被检测到, 位于48.81 cM处, 与mk1681紧密连锁, LOD值为4.2, 置信区间为8.65~8.85 Mb, 可解释7.0%的表型变异, 来自于02428的等位基因可推迟抽穗期1.7 d。只在E2环境中被检测到, 位于118.41 cM处, 与mk1725紧密连锁, LOD值为3.0, 置信区间为22.05~22.55 Mb, 可解释5.2%的表型变异, 来自于02428的等位基因可缩短抽穗期1.2 d。第8染色体上共有2个抽穗期QTL, 即和。在E1环境检测到的位置是42.41 cM, 与mk1823紧密连锁, LOD值为5.6, 置信区间为4.20~4.55 Mb, 可解释9.5%表型变异, 来自于02428的等位基因能推迟抽穗期2 d; 在E3环境检测到的位置是42.44 cM, 与mk1823紧密连锁, LOD值为3.9, 置信区间为4.20~4.55 Mb, 可解释7.5%表型变异, 来自于02428的等位基因能推迟抽穗期2.4 d。仅在E1环境被检测到, 位于56.71 cM处, 与mk1830紧密连锁, LOD值为4.3, 置信区间为4.65~5.15 Mb, 可解释7.4%表型变异, 来自于02428的等位基因能推迟抽穗期1.7 d。
在第9染色体上125.4 cM处, 仅在E4环境中被检测到, 与mk2090连锁。其LOD值为2.8, 置信区间为16.55~16.75 Mb, 表型贡献率为4.6%, 来自于02428的等位基因可缩短生育期1.0 d。
第10染色体56.71 cM处为, 仅在E1环境被检测到, 它与mk2245紧密连锁, LOD值为3.5, 置信区间为16.35~16.65 Mb, 可解释6.7%表型变异, 来自于02428的等位基因可缩短抽穗期1.6 d。同一染色体上的在E1、E2和E4环境被重复检测到, 置信区间为16.75~17.25 Mb, E1环境的LOD值为4.5, 贡献率为8.2%, 来自于02428的等位基因可缩短抽穗期1.4 d; E2环境的LOD值为3.2, 贡献率为5.4%, 来自于02428的等位基因可缩短抽穗期1.5 d; E4环境的LOD值为9.6, 贡献率为17.0%, 来自于02428的等位基因可缩短抽穗期1.7 d。第10染色体67.41 cM处是, 仅在E2环境中被检测到, LOD值为4.2, 可解释7.5%表型变异, 置信区间为17.75~17.95 Mb, 来自于02428的等位基因可缩短抽穗期1.4 d。
2.4 注释基因筛选
在3个环境中可重复检测到, 其置信区间最大时为28.15~28.65 Mb, 在该染色体区间有33个注释基因(The Rice Annotation Project Database), 逐一筛除发现有3个位点很可能影响抽穗期, 分别是、和其中,与拟南芥中()同源, 可影响水稻抽穗期、胚的发育及小花器官数目。和的基因产物是可表达的蛋白, 其涉及的生物学进程包括花器官的发育、生殖生长等。
DNA双向测序发现, 玉针香和02428的3个候选基因间均有序列差异(图3)。玉针香与02428的基因存在23处碱基差异, 共18处为单碱基的转换, 5处为3碱基以内的插入缺失; 其中7处在CDS区变异, 有5处发生错义突变, 分别位于CDS+830 bp (TTT/TCT)、CDS+1008 bp (GAG/GAT)、CDS+150 bp (TTG/TTT)、CDS+2413 bp (CGT/TGT)和CDS+2 418 bp (GAC/GAG)。玉针香与02428的基因存在23处碱基差异, 其中21处为单碱基的转换, 2处为单碱基插入缺失; 其中2处在CDS区的变异, 有1处为错义突变, 位置是CDS+394 bp (AAT/TAT)。玉针香与02428的基因存在13处碱基差异, 其中9处为单碱基的转换, 4处为插入缺失; 其中2处在CDS区变异, 有1处为错义突变, 位置是CDS+883 bp (CTT/TTT)。同时, 测序比对发现, 02428中3个候选基因的CDS序列与日本晴参考序列完全相同。
除此之外, 还在其他QTL的置信区间内找到一些与抽穗期相关可能性较大的注释基因, 它们都涉及到花的发育过程(表4)。
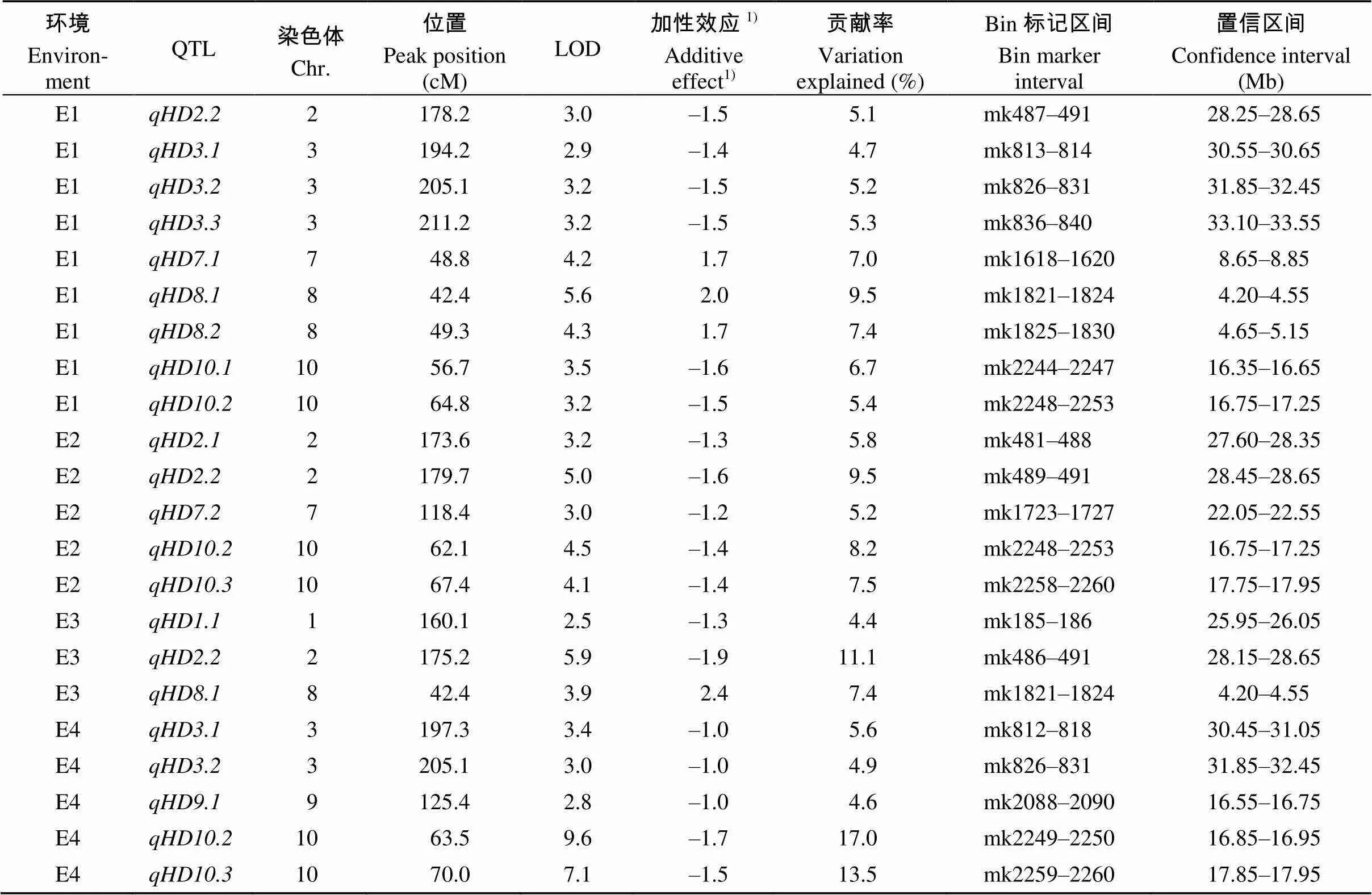
表3 4个环境下检测到的抽穗期QTL
1)指来自于02428的等位基因造成的加性效应, 正值为延长抽穗期, 负值为缩短抽穗期。
1)Additive effect from 02428’s allele. Positive (negative) values mean increasing (decreasing) heading date.
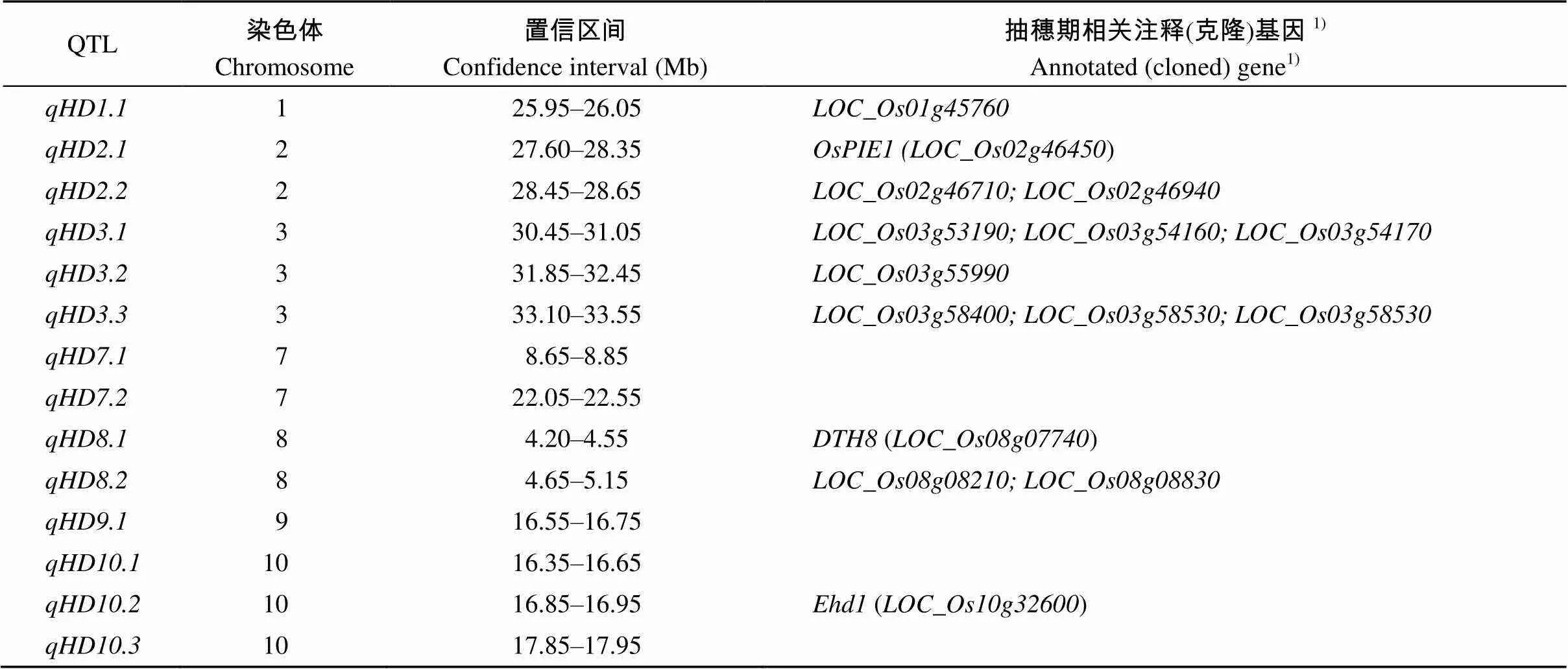
表4 QTL置信区间内注释基因筛选
1)Source: CHINA RICE DATA CENTER and The Rice Annotation Project Database.
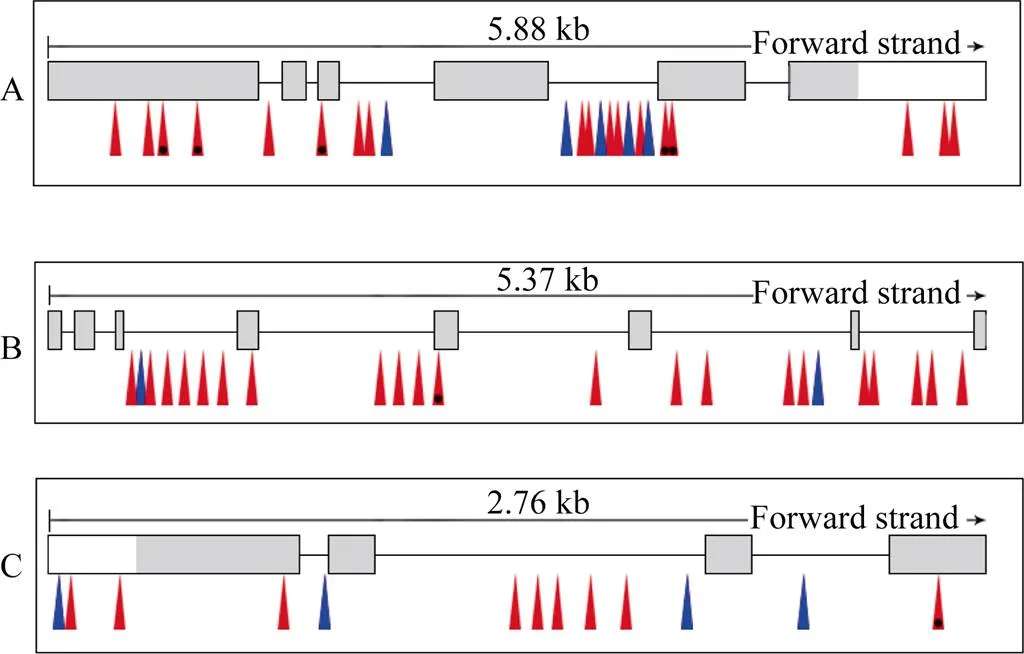
图3 玉针香与02428之间候选基因的结构和变异
A:; B:; C:。黑色框: 外显子; 灰色部分: 编码序列; 红色箭头: SNP; 带黑点红色箭头: 错义突变; 蓝色箭头: 插入缺失。
A:; B:; C:Frames with black lines: exon; Grey boxes: protein coding sequence; Red arrow: SNP; Red arrow with a black point: missense mutation; Blue arrow: InDel.
3 讨论
3.1 采用Bin图谱定位QTL的优势
目前大多数研究者采用传统分子标记(如SSR、RFLP等)构建遗传图谱[25], 精度约为1~10 Mb, 存在标记数量少且分布不均匀的问题, 影响了QTL定位的精确度, 另外标记密度较小还可能导致某些双交换位点的漏测[26]。Bin图谱与传统的遗传图谱相比, 标记密度高, 精度达100 kb, 且一个Bin内部包含多个不发生重组的SNP, 因而双交换能够被精确检测。同时, Bin图谱是基于测序技术构建的, 能提供准确的物理位置, 可使遗传分析和QTL定位更准确。此外, 测序分型得到的大量SNP标记可快速应用于分子育种。本研究采用GBS技术构建了以SNP为基础的高分辨率Bin图谱, 包含2711个Bin标记, 标记间平均物理距离137.36 kb, 标记整体的分布达到精细作图的密度, 可直接从定位区间筛选候选基因。另外, 定位结果显示, 在LOD值超过阈值的一段区域一般存在多个峰值, 可定位到多个QTL, 如位置相邻的和以及和等, 说明Bin图谱对QTL的检测更精细, 可将位置相邻的QTL有效地分离、解析。
3.2 本研究与前人定位QTL的比较
本研究定位的在第1染色体上, 仅在E3环境中被检测到, 其LOD值为2.5, 置信区间为25.95~26.05 Mb, 表型贡献率为4.5%, 在前人的研究中未见报道。
位于第2染色体27.95 Mb处, 置信区间为17.60~28.35 Mb, 表型贡献率为5.8%, LOD值为3.2, 在该区间有多篇相关的报道[27-32], 其中Thomson等[31]报道的表型贡献率为4.4%, LOD为4.06, 在5个环境中检测到1次, 与位置也最近, 它们可能为同一QTL。在第2染色体上, E1环境于28.55 Mb位置检测到1个QTL, 其置信区间为28.25~28.65 Mb; E2环境于28.65 Mb位置检测到1个QTL, 置信区间为28.45~28.65 Mb; E3环境于28.35 Mb位置检测到1个QTL, 置信区间为28.15~28.65 Mb。这3个QTL位置十分相近, 置信区间十分相似, 因此视为同一QTL, 命名为, 它在前人的研究中未见报道, 很可能是与水稻抽穗期相关的新位点。
第3染色体上检测到3个QTL和。在2个环境中被重复检测到, 在E1环境定位的位置是30.55 Mb, 置信区间分别为30.55~30.65 Mb; E4环境定位的位置是30.95 Mb, 置信区间为30.45~31.05 Mb。也在2个环境中被重复检测到, 定位的位置是32.35 Mb, 置信区间为31.85~32.45 Mb。仅在E1环境被检测到, 位于33.35 Mb, 置信区间为33.10~ 33.55 Mb。这3个QTL所在区域已有相关报道[31,33-34]。
位于第7染色体8.85 Mb处, 置信区间为8.65~8.85 Mb, 已精细定位的包含了该区间[35]。同一染色体上的位于22.35 Mb处, 置信区间为22.05~22.55 Mb, 该区间位于Thomson等[31]定位的所在区间RM125–RM336内部, Jiang等[36]定位的与的区间也有部分重叠。
位于第8染色体4.45 Mb处, 置信区间为4.20~ 4.55 Mb, 与已克隆的[39]位置相吻合, 都表现为长日照延长抽穗期。位于第8染色体5.15 Mb处, 置信区间为4.65~5.15 Mb, 在该区段也有多个相关报道[37-40]。
仅在E4环境被检测到, 位于第9染色体16.75 Mb处, 与mk2090紧密连锁, 置信区间为16.55~16.75 Mb,未发现相关报道。
位于第10染色体16.45 Mb处, 置信区间为16.35~16.65 Mb, 与Thomson等[31]定位的区间有部分相重叠。在第10染色体上, E1环境于17.25 Mb位置检测到1个QTL, 其置信区间为16.75~17.25 Mb; E2环境于16.75 Mb位置检测到1个QTL, 置信区间为16.75~ 17.25 Mb。这2个QTL位置相近、区间相同, 因此视为同一QTL, 命名为, Doi等[7]报道的的位置在其区间内。同在第10染色体上的位于17.85 Mb处, 置信区间为17.75~17.95 Mb, Mei等[32]报道了与抽穗期相关的RG241a–CDO98染色体区段在的区间内, Zhou等[28]报道的与区间基本相同。
3.3 检测到的QTL应用价值及研究价值
本研究在4个环境中共检测到14个影响抽穗期的QTL, 分布于第1、第2、第3、第7、第8、第9和第10染色体上。除、、、和能在2个及以上环境被检测到外, 其他QTL只在单个环境被检测到。其中,和所在的位置分别是克隆基因和。
Hori等[20]同时用12个群体进行抽穗期相关的QTL定位, 在其中3个群体中能检测到, 表型贡献率为7.9%~74.0%; 在其中2个群体中能检测到, 表型贡献率为18.2%~18.3%。Cheng等[2]同时用3个群体进行抽穗期相关的QTL定位, 在其中1个群体中能检测到, 表型贡献率为3.5%; 在3个群体中均能检测到, 表型贡献率为3.6%~33.7%。而本研究中检测到的表型贡献率为7.5%~9.5%;的表型贡献率为5.4%~ 17.0%。这些研究表明, 对于不同群体, 由于材料遗传背景影响, QTL贡献率的变化很大; 其次, 图谱的精度对QTL贡献率的估算影响也较大, 传统分子标记构建的图谱精度通常约为4 Mb, 两标记之间经常会存在多个相关基因或QTL簇, 粗定位到的QTL贡献率会较高。因此我们认为, 重点关注QTL在不同环境或不同群体中的重复定位情况对聚合育种的意义更大。
和在定位到的QTL中表型贡献率相对较小, 表达也较不稳定, 只能2次被重复检测到, 因而利用的可能性小。在两年的早造中被重复检测到, 仅在早造环境特异表达, 表现为延长生育期, 这些都与在长日条件下延长抽穗期的特性相对应, 它对于将早熟品种改良成半晚熟的高产品种具有一定价值。和都能在3个环境中被重复检测到, 且都能够缩短生育期, 说明它们受环境影响较小、表达较稳定, 对光照和温度变化不敏感, 它们的表型贡献率最低时分别为5.1%和5.4%, 表型贡献率最高时分别为11.2%和17.0%, 因此对于QTL聚合改良品种生育期具有重要价值。
此外,的置信区间在3次定位中略有变化, 在E2环境判定的置信区间较其他2个环境中小,不在此置信区间内, 但此时对表型的贡献率仍然达到9.5%。因此和更有可能是导致抽穗期表型变异的新QTL, 具体情况还需深入研究。
[1] Fujino K, Sekiguchi H. Mapping of quantitative trait loci controlling heading date among rice cultivars in the northernmost region of Japan., 2008, 58: 367–373
[2] Cheng L R, Wang J M, Ye G, Luo C G, Xu J L, Li Z K. Identification of stably expressed QTL for heading date using reciprocal introgression line and recombinant inbred line populations in rice., 2012, 94: 245–253
[3] 戴高兴, 杨占烈, 邓国富, 张迎信, 王会民, 翟荣荣, 曹立勇, 程式华. 超级杂交稻协优9308重组自交系主茎叶片数的动态QTL分析. 中国水稻科学, 2012, 26: 291–296 Dai G X, Yang Z L, Deng G F, Zhang Y X, Wang H M, Zhai R R, Cao L Y, Cheng S H. QTL analysis for leaf number on main stem in RILs of super hybrid rice Xieyou 9308.i, 2012, 26: 291–296 (in Chinese with English abstract)
[4] Yano M, Katayose Y, Ashikari M, Yamanouchi U, Monna L, Fuse T, Baba T, Yamamoto K, Umehara Y, Nagamura Y. Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene., 2000, 12: 2473–2484
[5] Takahashi Y, Shomura A, Sasaki T, Yano M. Hd6, a rice quantitative trait locus involved in photoperiod sensitivity, encodes the α subunit of protein kinase CK2., 2001, 98: 7922–7927
[6] Kojima S, Takahashi Y, Kobayashi Y, Monna L, Sasaki T, Araki T, Yano M. Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions., 2002, 43: 1096–1105
[7] Doi K, Izawa T, Fuse T, Yamanouchi U, Kubo T, Shimatani Z, Yano M, Yoshimura A., a B-type response regulator in rice, confers short-day promotion of flowering and controlsgene expression independently of Hd1., 2004, 18: 926–936
[8] Xue W, Xing Y, Weng X, Zhao Y, Tang W, Wang L, Zhou H, Yu S, Xu C, Li X, Zhang Q. Natural variation inis an important regulator of heading date and yield potential in rice., 2008, 40: 761–767
[9] Wei X, Xu J, Guo H, Jiang L, Chen S, Yu C, Zhou Z, Hu P, Zhai H, Wan J. DTH8 suppresses flowering in rice, influencing plant height and yield potential simultaneously., 2010, 153: 1747–1758
[10] Bian X F, Liu X, Zhao Z G, Jiang L, Gao H, Zhang Y H, Zheng M, Chen L M, Liu S J, Zhai H Q. Heading date gene,controlled late flowering inSteud. by down-regulating., 2011, 30: 2243–2254
[11] Yan W H, Wang P, Chen H X, Zhou H J, Li Q P, Wang C R, Ding Z H, Zhang Y S, Yu S B, Xing Y Z. A major QTL, Ghd8, plays pleiotropic roles in regulating grain productivity, plant height and heading date in rice., 2011, 4: 319–330
[12] Matsubara K, Ogisotanaka E, Hori K, Ebana K, Ando T, Yano M. Natural variation in Hd17, a homolog of Arabidopsis ELF3 that is involved in rice photoperiodic flowering., 2012, 53: 709–716
[13] Gao H, Zheng X, Fei G, Chen J, Jin M, Ren Y, Wu W, Zhou K, Sheng P, Zhou F, Jiang L, Wang J, Zhang X, Guo X, Wang J, Cheng Z, Wu C, Wang H, Wan J. Ehd4 encodes a novel and Oryza-Genus-Specific regulator of photoperiodic flowering in rice.2013, 9: e1003281
[14] Hori K, Ogisotanaka E, Matsubara K, Yamanouchi U, Ebana K, Yano M., a gene for casein kinase I, is involved in the control of rice flowering time by modulating the day-length response., 2013, 76: 36–46
[15] Koo B H, Yoo S C, Park J W, Kwon C T, Lee B D, An G, Zhang Z Y, Li J J, Li Z C, Paek N C. Natural variation in OsPRR37 regulates heading date and contributes to rice cultivation at a wide range of latitudes., 2013, 6: 1877–1888
[16] Ogisotanaka E, Matsubara K, Yamamoto S, Nonoue Y, Wu J, Fujisawa H, Ishikubo H, Tanaka T, Ando T, Matsumoto T. Natural variation of thecontributes to flowering time divergence in rice., 2013, 8: e75959
[17] Wu W, Zheng X M, Lu G, Zhong Z, Gao H, Chen L, Wu C, Wang H J, Wang Q, Zhou K, Wang J, Wu F, Zhang X, Guo X, Cheng Z, Lei C, Lin Q, Jiang L, Wang H, Ge S, Wan J. Association of functional nucleotide polymorphisms at DTH2 with the northward expansion of rice cultivation in Asia., 2013, 110: 2775–2780
[18] Sun B, Zhan X D, Lin Z C, Wu W X, Yu P, Zhang Y X, Sun L P, Cao L Y, Cheng S H. Fine mapping and candidate gene analysis of qHD5, a novel major QTL with pleiotropism for yield-related traits in rice (L.).2016, 130: 1–12
[19] Shen G, Xing Y. Two novel QTLs for heading date are identified using a set of chromosome segment substitution lines in rice (L.)., 2014, 41: 659–662
[20] Hori K, Nonoue Y, Ono N, Shibaya T, Ebana K, Matsubara K, Ogisotanaka E, Tanabata T, Sugimoto K, Taguchishiobara F. Genetic architecture of variation in heading date among Asian rice accessions., 2015, 15: 115
[21] Xie W, Feng Q, Yu H, Huang X, Zhao Q, Xing Y, Yu S, Han B, Zhang Q. Parent-independent genotyping for constructing an ultrahigh-density linkage map based on population sequencing., 2010, 107: 10578–10583
[22] Yu H, Xie W, Wang J, Xing Y, Xu C, Li X, Xiao J, Zhang Q. Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers.e, 2012, 6: e17595
[23] Chen L, Gao W, Guo T, Huang C, Huang M, Wang J, Xiao W, Yang G, Liu Y, Wang H, Chen Z. A genotyping platform assembled with high-throughput DNA extraction, codominant functional markers, and automated CE system to accelerate marker-assisted improvement of rice., 2016, 36: 123, https://doi.org/10.1007/s11032-016-0547-y
[24] McCouch S R. Gene nomenclature system for rice., 2008, 1: 72–84
[25] Golicz A A, Bayer P E, Edwards D. Skim-based genotyping by sequencing., 2015, 1245: 257–270
[26] Yu H, Xie W, Li J, Zhou F, Zhang Q. A whole-genome SNP array (RICE6K) for genomic breeding in rice.2014, 12: 28–37
[27] Zou J H, Pan X B, Chen Z X, Xu J Y, Lu J F, Zhai W X, Zhu L H. Mapping quantitative trait loci controlling sheath blight resistance in two rice cultivars (L.).2000, 101: 569–573
[28] Zhou Y, Li W, Wu W, Chen Q, Mao D, Worland A J. Genetic dissection of heading time and its components in rice., 2001, 102: 1236–1242
[29] Li Z K, Yu S B, Lafitte H R, Huang N, Courtois B, Hittalmani S, Vijayakumar C H M, Liu G F, Wang G C, Shashidhar H E, Zhuang J Y, Zheng K L, Singh V P, Sidhu J S, Srivantaneeyakul S, Khush G S. QTL×environment interactions in rice: I. Heading date and plant height., 2003, 108: 141–153
[30] Xiao J, Li J, Yuan L, Tanksley S D. Identification of QTLs affecting traits of agronomic importance in a recombinant inbred population derived from a subspecific rice cross., 1996, 92: 230–244
[31] Thomson M J, Tai T H, Mcclung A M, Lai X H, Hinga M E, Lobos K B, Xu Y, Martinez C P, McCouch S R. Mapping quantitative trait loci for yield, yield components and morphological traits in an advanced backcross population betweenand thecultivar Jefferson., 2003, 107: 479–493
[32] Mei H W, Luo L J, Ying C S, Wang Y P, Yu X Q, Guo L B, Paterson A H, Li Z K. Gene actions of QTLs affecting several agronomic traits resolved in a recombinant inbred rice population and two testcross populations., 2003, 107: 89–101
[33] Takeuchi Y, Hayasaka H, Chiba B, Tanaka I, Shimano T, Yamagishi M, Nagano K, Sasaki T, Yano M. Mapping quantitative trait loci controlling cool-temperature tolerance at booting stage in temperate japonica rice., 2001, 51: 191–197
[34] Sarma R N, Gill B S, Sasaki T, Galiba G, Sutka J, Laurie D A, Snape J W. Comparative mapping of the wheat chromosome 5A Vrn-A1 region with rice and its relationship to QTL for flowering time., 1998, 97: 103–109
[35] Lin H, Liang Z W, Sasaki T, Yano M. Fine mapping and characterization of quantitative trait loci Hd4 and Hd5 controlling heading date in rice., 2003, 53: 51–59
[36] Jiang L, Xu J, Wei X, Wang S, Tang J, Zhai H, Wan J. The inheritance of early heading in the rice variety USSR5., 2007, 34: 46–55
[37] 国广泰史, 钱前, 佐藤宏之, 滕胜, 曾大力, 藤本宽, 朱立煌. 水稻纹枯病抗性QTL分析. 遗传学报, 2002, 29: 50–55 Kunihiro Y, Qian Q, Sato H, Teng S, Zeng D L, Fujimoto K, Zhu L H. QTL analysis of sheath blight resistance in rice (L.)., 2002, 29: 50–55 (in Chinese with English abstract)
[38] Wang C M, Yasui H, Yoshimura A, Wan J M, Zhai H Q. Identification of quantitative trait loci controlling F2sterility and heading date in rice., 2002, 29: 339–342
[39] Lu C, Shen L, Tan Z, Xu Y, He P, Chen Y, Zhu L. Comparative mapping of QTLs for agronomic traits of rice across environments using a doubled haploid population., 1996, 93: 1211–1217
[40] 谭震波, 沈利爽, 况浩池, 陆朝福, 陈英, 周开达, 朱立煌. 水稻上部节间长度等数量性状基因的定位及其遗传效应分析. 遗传学报, 1996, 23: 439–446Tan Z B, Shen L S, Kuang H C, Lu C F, Chen Y, Zhou K D, Zhu L H. Identification of QTLs for lengths of the top internodes and other traits in rice and analysis of their genetic effects., 1996, 23: 439–446 (in Chinese with English abstract)
QTL Mapping for Heading Date in Rice Using High-density Bin Map
DONG Ji-Chi**, YANG Jing**, GUO Tao, CHEN Li-Kai, CHEN Zhi-Qiang*, and WANG Hui*
National Engineering Research Centre of Plant Space Breeding / South China Agricultural University, Guangzhou 510642, Guangdong, China
A recombination inbred lines (RIL) population including 192 lines derived from an inter-subspecific cross betweenrice ‘Yuzhenxiang’ andrice ‘02428’ was used in the experiment. The two parent varieties and RIL population were separately sequenced by Whole Genome Sequencing (WGS) and Genotyping-By-Sequencing (GBS) to construct a genetic linkage map with 2711 recombination Bin markers. The number of markers on 12 chromosomes ranged from 162 to 311, and the average physical distance between two markers was 137.68 kb. The WinQTL Cartographer 2.5 was used to analysis QTLs associated with heading date in four different environments. A total of 14 QTLs associated with heading date were detected on chromosomes 1, 2, 3, 7, 8, 9, and 10. Among them,and, which explained 5.14%-11.15% and 5.35%-16.97% of the total phenotypic variation for heading date separately, could be detected in three environments and shorting heading date about 1.66 days and 1.56 days on average. Thetwo QTLs could inherit stablely, having a good potential to be applied in QTL pyramiding. After comparing the physical positions of these QTLs with those previously reported, we found 11 QTLs were located in the same or near position, among them,,andwere newly reported. Furthermore, we found one cloned geneand two annotated genesandin the genomic region of, might be related to heading date. DNA sequence comparison between YZX and 02428 revealed that all the three genes could be candidate genes.
rice; heading date; Bin map; QTL mapping
2017-12-14;
2018-03-25;
2018-04-16.
10.3724/SP.J.1006.2018.00938
王慧, E-mail: wanghui@scau.edu.cn; 陈志强, E-mail: chenlin@scau.edu.cn
**同等贡献(Contributed equally to this work)
董骥驰, E-mail: Johnkeatsno.1@gmail.com; 杨靖, E-mail: 1349643559@qq.com
URL: http://kns.cnki.net/kcms/detail/11.1809.S.20180416.0843.004.html
本研究由国家现代农业产业技术体系建设专项(CARS-01-12), 国家重点研发计划项目(2016YFD0102102)和广东省应用型研发项目(2015B020231011)资助。
This study was supported by the China Agriculture Research System (CARS-01-12), the National Key Research and Development Program of China (2016YFD0102102), and the Research and Development Program for Application in Guangdong Province (2015B020231011).
