靖安白茶芽和叶的转录组数据组装及基因功能注释
2018-06-11李明玺王敏甘玉迪刘洋黄莹捷万春鹏
李明玺,王敏,甘玉迪,刘洋,黄莹捷,万春鹏
(1.江西农业大学农学院,江西农业大学茶学与茶文化研究中心,江西南昌 330045)(2.南京农业大学园艺学院,江苏南京 210095)
白茶,一种是按照制作工艺命名的白茶,通过不揉不炒,自然萎凋的独特加工工艺制成的六大基本茶类之一轻萎凋茶类[1]。另一种是按照茶树品种如安吉白茶、溧阳白茶、福鼎大白茶与政和大白茶等命名的品种名。白茶除了饮用外,还可以作为药用使用,其性清凉,能退热、降火,可治麻疹[2],现代研究表明白茶具有很好的抗氧化、抗肿瘤、降血糖、减肥降脂和保肝护肝等作用[3]。白茶富含茶多酚和茶氨酸等活性成分,茶叶多酚的主要成分有儿茶素(黄烷醇)类、酚酸类、黄酮醇类和花青素等。其中黄酮醇类是一种低分子量的茶多酚类物质,主要以黄酮糖苷的形式存在,少部分以苷元形式存在,占干物质含量 2~4%,目前已知的植物黄酮类物质巳有8000多种[4]。茶叶中黄酮醇类化合物的含量与茶树品种、生长季节、加工方式和贮藏环境等密切相关,黄酮醇苷类化合物是茶汤中主要的涩味物质,也是构成汤色色泽的重要因子,对人体具有预防心血管疾病、抗氧化、保护肝脏及防癌等作用,是目前备受关注的茶叶天然产物之一[5~10]。茶氨酸,茶树中一种比较特殊的氨基酸,茶氨酸一般占茶叶干物质总重的 1~2%,不参与蛋白质的合成,以游离形式存在,占茶叶中游离氨基酸总量的50%左右[11]。自然界存在的茶氨酸为L型,系统命名为:N-乙基-γ-谷氨酰胺,极易溶于水。茶氨酸是茶叶鲜爽味的主要成分,并且可缓解咖啡因的苦味和茶多酚的苦涩味,对人体具有神经保护[12]、抗肥胖[13]、降血压[14]、促进认知能力[15]、抗肿瘤、改善记忆和抗疲劳等功效[16]。
随着高通量测序技术的出现,由于该技术能快速地全面获取研究对象在某一状态下基因转录信息,因此该技术广泛的应用生物体转录组基因表达分析,能够准确发掘重要功能基因[17,18]。近年来人们因白茶具有一定的药用作用以及潜在的一些保健功能,对白茶的关注度逐渐增加。先前已有相关的报道对白茶的品种、制作工艺、品质、化学成分和药理活性等进行研究[19,20],但是对于白茶的芽叶中相关基因如何调控茶多酚和茶氨酸等内含成分代谢尚未见报道。因此本研究通过高通量测序技术,对白茶芽叶相关代谢途径进行研究,探索白茶芽叶中类黄酮及谷胱甘肽代谢的相关基因调控对应的代谢途径。
1 材料与方法
1.1 材料
供试植物于2017年5月10日采集于江西省宜春市靖安县罗湾乡白云白茶有限公司茶园,样品采集当天及前五天均为晴天。茶树品种:Camellia sinensiscultivar Jing'anbaicha,8年树龄,海拔780米红壤山地丘陵,属于北亚热带湿润气候,春季回暖迟,有春寒,夏季炎热时间长,秋季凉得快,冬季较寒冷,四季分明。气候温和,平均气温在15.0度左右,要比平原低3度以上,该地白茶发芽平均比平地晚10 d左右。
1.2 试验方法
1.2.1 RNA提取、文库构建与检测
分别从3份白茶芽、叶样品中采用TRIzol法提取总RNA[21],并对RNA浓度、纯度、完整性进行检测以确保数据的准确性。样品检测合格后,用带有Oligo(dT)的磁珠富集真核生物mRNA。随后加入。随后加入fragmentation buffer将mRNA打断成短片段,以打断后的mRNA为模板合成一链cDNA,然后合成二链cDNA。随之利用 AMPure XP beads将合成的二链cDNA纯化,进行末端修复、加A尾并连接测序接头,再用AMPure XP beads进行片段大小选择。最后进行PCR扩增,纯化PCR产物,得到最终文库。文库构建完成后,先使用Qubit 2.0进行初步定量,随后使用Agilent 2100对文库的插入片段大小进行检测,使用Q-PCR方法对文库的有效浓度进行准确定量,质检合格后使用Illumina 4000高通量测序平台(HiSeq/MiSeq)进行测序。
1.2.2 转录组测序结果组装、拼接
由Illumina 4000高通量测序平台测序所得的数据称为raw reads或raw data。由于原始数据存在低质量、可能的接头等序列,为了确保分析结果的可靠准确,需要先将这部分序列去除才能用于下一步的分析,使用FastQC[22]对原始数据进行质控,以确定测序数据是否适用于后续分析。随后进行Trinity[23]进行转录本的拼接,得到的转录本序列。利用 CD-HIT[24]将所有拼接出来的转录本进行启发式聚类,选择每一类中最长的转录本后去除其余冗余序列,从而构建非冗余的转录本集合,此集合即为表达基因集合。最后从CD-HIT处理后序列挑选出最长的转录本作为unigene。
1.2.3 基因功能注释
对获得的转录组序列同常用的几种数据库进行比对注释,其中主要的数据库包括:Nr(NCBI蛋白质非冗余数据库),Pfam(蛋白家族数据库),KOG(真核生物直系同源基因数据库),Swiss-Prot(经实验验证的蛋白序列数据库),KEGG(基因产物、细胞中代谢途径以及基因产物功能的数据库),GO(基因本体数据库)。
1.2.4 基因表达水平分析
RSEM[25]对bowtie2[26]的比对结果进行统计,进一步得到了每个样品比对到每个基因上的 reads数目,并将其转换成FPKM[27]值,进而分析基因的表达水平。
2 结果与分析
2.1 测序结果与Trinity拼接
白茶芽转录组共得到 56882837 raw reads,56456634 clean reads,Q20值为 94.56%,Q30值为87.32%,序列碱基GC含量为44.84%;叶转录组共得到 58092923 raw reads,57800442 clean reads,Q20 值为94.51%,Q30值为87.32%,序列碱基GC含量为44.54%;表明白茶转录组测序质量较高,为随后的数据组装拼接,分析研究提供了较好的数据基础。经过Trinity转录本拼接和CD-HIT去冗余序列处理结果见表1。

图1 拼接后unigene长度分布频数图Fig.1 Length distribution of all unigene after splicing
对拼接得到的转录本序列和 unigene序列长度分布统计可知长度<300的最多,频数31294(如图1所示)。以上数据表明转录本拼接较好,利于之后的分析研究。

表1 不同处理后序列数量、N50长度和总碱基数统计表Table 1 Sequence number, length of N50 and total bases treated by different data processing
2.2 基因功能注释结果
对获得的转录组序列进行数据库比对注释可知,共注释了unigene数量为100568,Nr(NCBI蛋白质非冗余数据库)注释的unigene数量为32530,占比最多为32.35%,KOG(真核生物直系同源基因数据库)注释的unigene数量为30087,占比29.82%,GO(基因本体数据库)注释的 unigene数量为 29010,占比28.84%,Swiss-Prot(经实验验证的蛋白序列数据库)注释的unigene数量为24553,占比24.41%,Pfam(蛋白家族数据库)注释的 unigene数量为 18576,占比18.47%(如表2所示)。

表2 Unigenes注释结果统计Table 2 Annotation statistics for all unigenes
2.2.1 Nr注释结果

图2 注释到NR数据库中主要物种分布图Fig.2 Distribution of the main species annotated to NR database
通过与 Nr库进行比对注释,可以获取本物种基因序列与近缘物种基因序列的相似性以及本物种基因的功能信息。将白茶的芽和叶与其他物种进行相似性比较,其中相似性排前十的有葡萄,相似度为27.95%;咖啡,相似度为5.77%;莲,相似度为4.8%;芝麻,相似度为4.54%;麻风树,相似度为3.59%;美花烟草,相似度为3.1%;甜橙,相似度为2.63%;梅,相似度为2.61%;烟草,相似度为2.52%;毛果杨,相似度为2.44%(如图2所示)。
2.2.2 GO分类

图3 GO分类图Fig.3 GO functional annotation distribution
对 unigene进行 GO注释之后,将注释成功的unigene按照GO三个大类的下一层级进行分类(如图3所示),包括生物学过程(Biological process,BP)、细胞组分(Cellular component,CC)和分子功能(Molecular Function,MF)。
按注释成功unigene多少统计,生物学过程中较多unigene注释的过程有代谢过程7237个、氧化还原过程3279个、生物过程3203个、磷酸化作用3095个;分子功能中较多unigene注释的分子功能有转移酶活性5227个、三磷酸腺苷结合体5186个、核苷酸结合域4933个、水解酶活性4276个、金属离子结合功能的4287个;细胞组分较多unigene注释的细胞组分有膜物质5108个、膜的有机组成部分4322个、细胞核3676个。
2.2.3 KOG分类
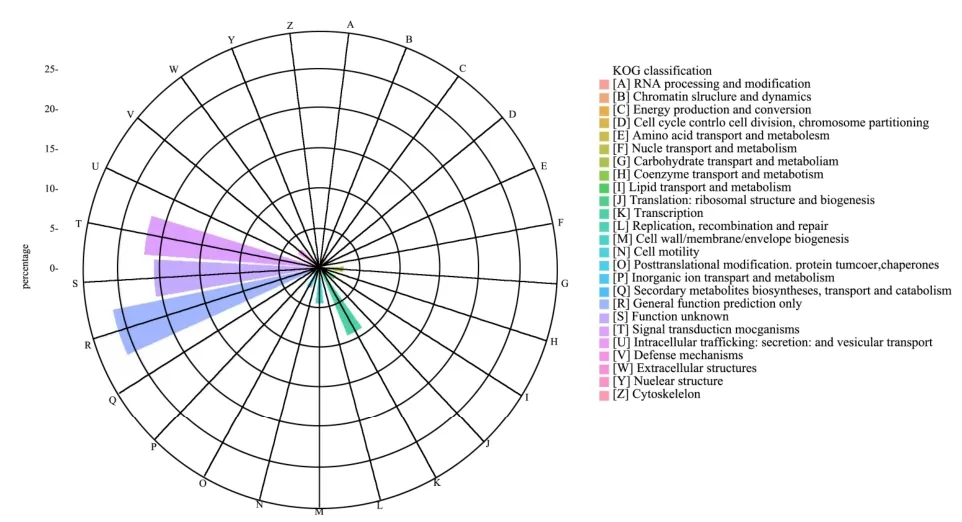
图4 KOG分类图Fig.4 KOG functional annotation distribution
2.2.4 KEGG分类
KOG (eukaryotic ortholog groups)注释是将真核生物的基因产物进行直系同源分类的数据库。将所有的unigene进行KOG注释分类,结果有6235条unigene被注释到25个功能类别中。其中一般功能预测注释到的unigene最多,有1640条;其次是,信号传导机制功能,被注释到的unigene共有1367条、未知功能注释到1289条;转录功能注释到unigene 571条,细胞壁/膜/包膜生物合成注释到 280条;防御机制注释到202条;碳水化合物转运代谢注释到187条(如图4所示)。
KEGG数据库涉及系统信息、基因组信息和化学信息,是系统分析基因产物在细胞中的代谢途径以及基因产物功能的数据库。利用KEGG数据库可以进一步研究基因在生物学上的复杂行为,有助于进一步研究特定基因的生物功能。将白茶芽叶转录组 unigene序列与KEGG数据库进行比对,共获得21001个注释结果,涉及的代谢通路可分为5大类(如图5所示):有机系统:4734条,22.54%,代谢7970条,37.95%,细胞过程2213条,10.54%,遗传信息处理3089条,14.71%,环境信息处理2995条,14.26%。其中环境信息处理中信号传导注释的unigene数最多,为2877条,其次,代谢过程中碳水化合物代谢注释的unigene为1643个,脂质代谢860个,能量代谢822个,氨基酸代谢1015个,辅助因子和维生素代谢346个,其他氨基酸代谢345个,萜类化合物和聚酮化合物代谢280个,糖链生物合成和代谢265个,其他次生代谢物生物合成574个,异生素生物降解和代谢364个,核苷酸代谢400个。
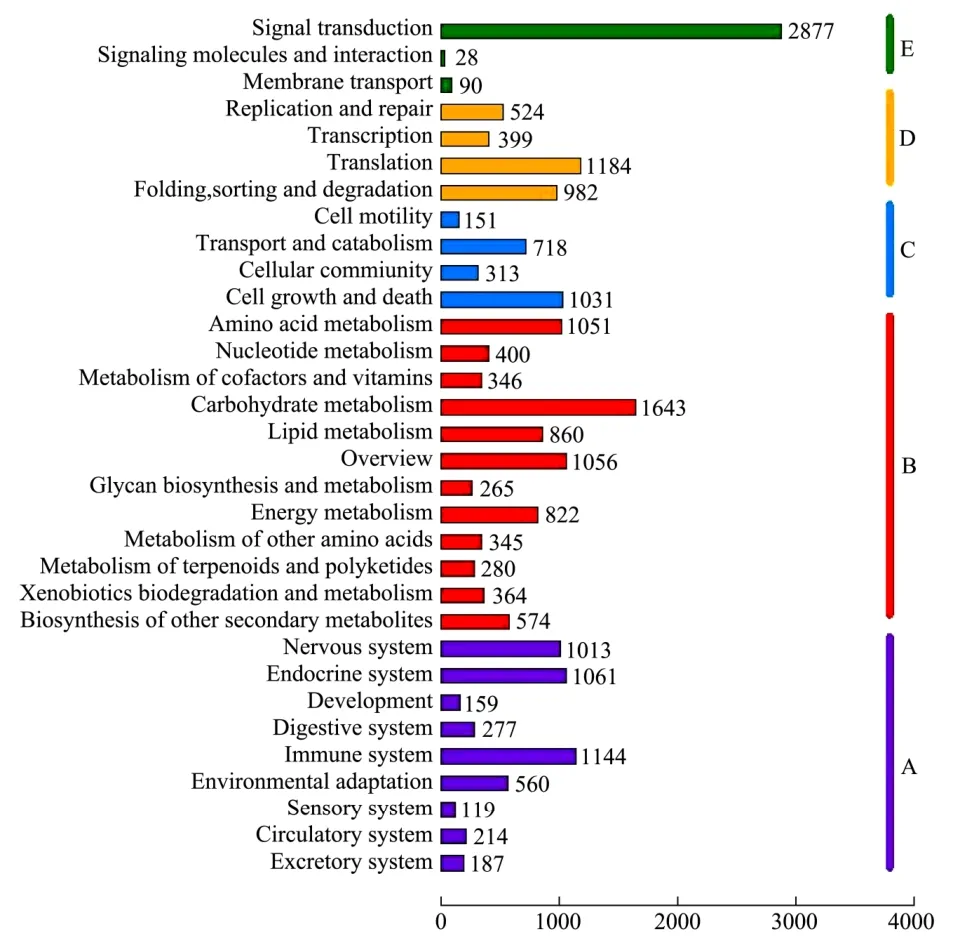
图5 KEGG代谢通路Fig.5 KEGG metabolic pathway
2.3 基因表达水平分析
2.3.1 基因表达水平统计

图6 白茶芽叶样品间相关性系数热图Fig.6 Correlation coefficient between buds and leaves of white tea
RSEM对bowtie2的比对结果进行统计,进一步得到了每个样品比对到每个基因上的 reads数目,并将其转换成FPKM值,进而分析基因的表达水平。对样品间基因表达水平相关性进行统计分析(如图6所示),芽的3个样本重复系数接近1,表明芽样本之间表达模式的相似度较高;叶的3个样本重复系数也接近 1,表明叶样本之间的表达模式的相似度也较高。而样本芽和叶之间的重复系数比较低,表明样品芽和叶之间的表达模式的相似度不高。因此,样本的选取是有意义的。
2.3.2 基因表达差异分析
对于有生物学重复的样品,采用edgeR[28]进行分析,上下调(芽vs叶)是从所有差异基因中过滤得到的过滤的值为fc为1.5,fdr 0.05(即只挑选变化倍数大于1.5,q值校正后小于0.05的基因),进行差异基因在不同样品间的聚类分析。据统计结果可知大部分上调基因位于芽中,而在芽中上调的基因在叶中是下调的(如图7所示)。

图7 unigene在不同样本间层次聚类热图Fig.7 Hierarchical clustering of unigene between buds and leaves of white tea
其中行表示的是每个 unigene表达的相对表达水平,列表示每个样本。其中表达值(FPKM)是经过Log2转化。
2.3.3 差异基因GO富集分析
将所有差异表达基因映射到Gene Ontology数据库的各个功能分类中,计算每个分类的基因数目,找出与整体基因组背景相比,在差异表达基因中显著富集的功能分类。
其中将上调基因进行富集分析(如图8所示),在细胞成分中,上调基因富集效果较明显的功能有,蛋白质-DNA复合物,GeneRatio占比 3.31%;微管,GeneRatio占比5.62%;DNA包装复合体,GeneRatio占比2.91%。分子功能中上调基因富集效果较明显的功能有核酸结合转录因子活性,GeneRatio占比9.93%;转录因子活性,GeneRatio占比9.93%;序列特异性DNA结合,GeneRatio占比2.46%;微管运动活动,GeneRatio占比2.96%。生物过程中上调基因富集效果较明显的功能有细胞增殖,GeneRatio占比4.86%;细胞周期过程,GeneRatio占比9.14%;有丝分裂细胞周期过程,GeneRatio占比6.65%;因此结果表明差异基因在细胞成分、分子功能、生物过程三方面主要调节物质的合成代谢。
而下调基因富集分析(如图9所示),细胞成分功能中富集较为明显的是类囊体,GeneRatio占比11.07%;光合膜,GeneRatio占比9%;叶绿体类囊体,GeneRatio占比9.17%;分子成分功能中富集较为明显的是氧化还原酶活性,GeneRatio占比22.31%;碳水化合物结合,GeneRatio占比5.32%;单加氧酶活性,GeneRatio占比3.47%。生物过程功能中富集较为明显的是光合作用,GeneRatio占比6.34%;光合作用,光反应,GeneRatio占比4.55%;氧化还原过程,GeneRatio占比22.2%。因此,下调基因在细胞成分、分子功能、生物过程三方面主要调节光合作用。
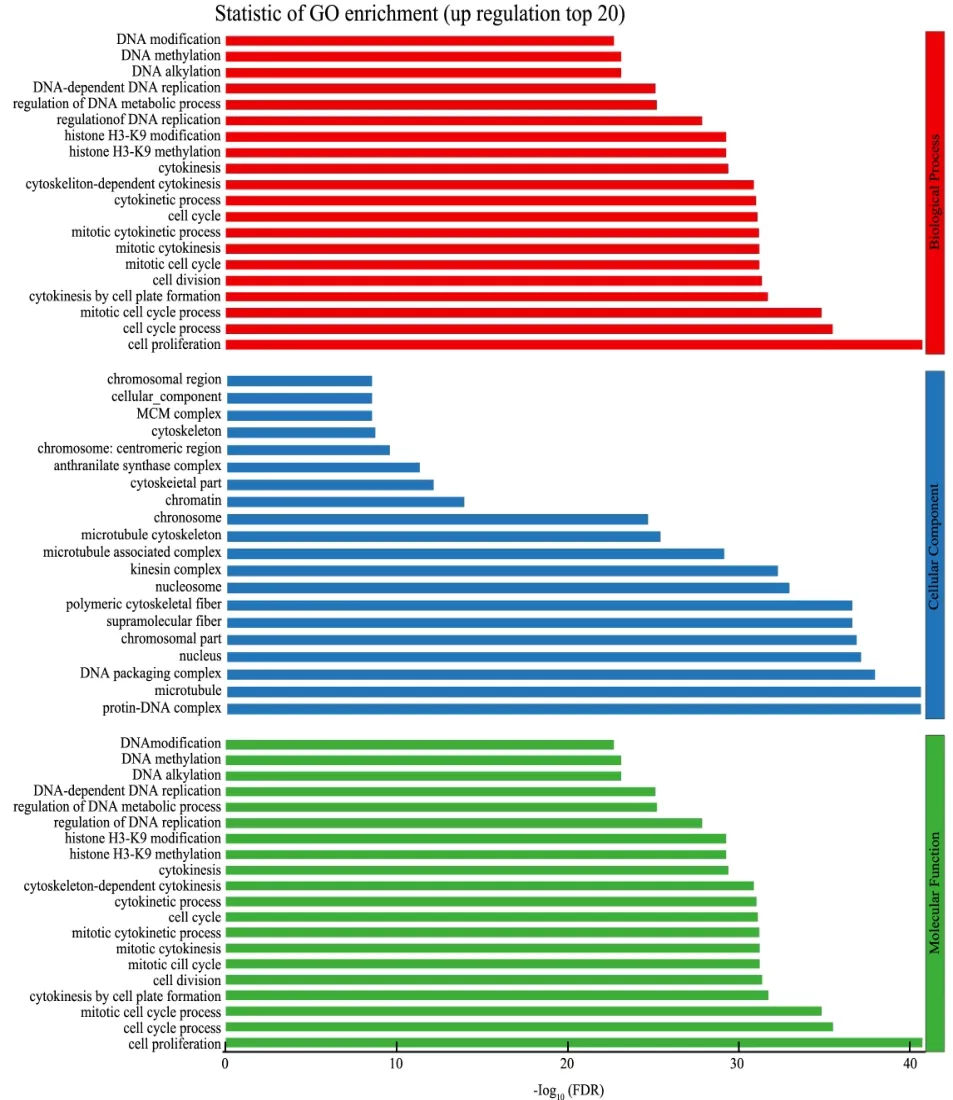
图8 上调差异基因(芽vs叶)的GO富集结果Fig.8 GO enrichment of up-regulation differential genes between buds and leaves
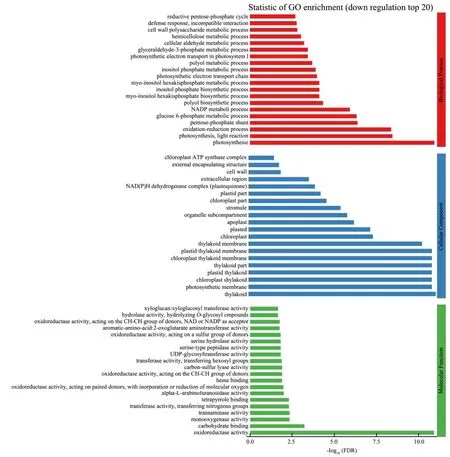
图9 下调差异基因(芽vs叶)的GO富集结果Fig.9 GO enrichment of down-regulation differential genes between buds and leaves
2.3.4 差异基因KEGG富集分析
Pathway显著性富集分析以KEGG Pathway为单位,应用超几何检验,找出差异基因相对于所有注释的基因显著富集的 Pathway。上调差异基因显著富集的通路有DNA复制、植物激素信号转导、黄酮类化合物的生物合成、淀粉与蔗糖代谢、芪类化合物,二芳基庚酸类,姜辣素的生物合成、苯丙素的生物合成、嘧啶代谢、半乳糖代谢、半胱氨酸和蛋氨酸代谢、谷胱甘肽代谢、苯丙氨酸、酪氨酸和色氨酸生物合成、类胡萝卜素的生物合成等(如图10所示)。
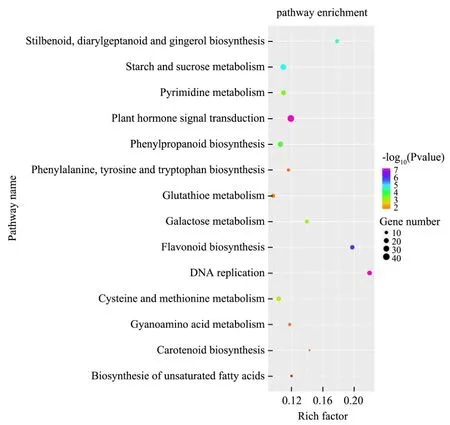
图10 上调基因(芽vs叶)参与的KEGG代谢通路富集结果Fig.10 Enrichment of up-regulation gene assignments to KEGG metabolic pathway

图11 上调基因参与的茶多酚生物合成通路Fig.11 Up-regulation gene assignments to tea polyphenol biosynthesis metabolic pathway

图12 下调基因(芽vs叶)参与的KEGG代谢通路富集结果Fig.12 Enrichment of down-regulation gene assignments to KEGG metabolic pathway
上调差异基因c18160_g1(Flavanone 4-reductase,EC:1.1.1.219 和 1.1.1.234);c5373_g1、c8716_g1、c4214_g1和c11557_g1(Leucoanthocyanidin reductase,EC:1.17.1.3);c19353_g1;c58885_g1;c34005_g1;c20606_g1、c11632_g1;c19095_g3;c2214_g1;c20311_g4;c15445_g2、c15445_g3(Shikimate O-hydroxycinnamoyl transferase,EC:2.3.1.133);c18003_g1(Leucoanthocy-anidin dioxygenase,EC:1.14.11.19)主要调节黄酮类化合物的生物合成,黄酮类化合物的生物合成与茶叶中的茶多酚形成密切相关,最终合成儿茶素、表儿茶素和没食子儿茶素等茶多酚(图11)。上调差异基c18645_g2;c18645_g1;c24761_g1;c9116_g1;c52277_g1;c37605_g1;c5153_g1;c47045_g1;c11055_g1;c20096_g7;c18271_g1;c13851_g1;c18630_g6调节谷胱甘肽代谢,谷胱甘肽是一种含Y-酰胺键和巯基的三肽且其中包含谷氨酸,与茶叶中的茶氨酸合成相关。
下调差异基因显著富集的通路有光合作用、乙醛酸盐代谢、光合生物固碳、苯丙素的生物合成、单萜类生物合成、碳代谢、半乳糖代谢、戊糖、葡萄糖醛酸转换、类胡萝卜素的生物合成、自噬调节、其他类型的O-聚糖的生物合成、氮代谢和甘油酯代谢等(如图12所示)。
下调差异基因c10462_g3;c15881_g1;c15881_g2;c8421_g1;c19569_g1调节类胡萝卜素的生物合成,类胡萝卜素的生物合成与茶树萜类和聚酮化合物形成有关,因此有利于叶绿素的形成,从而促进光合作用有关。
3 结论
3.1 Illumina 4000高通量测序平台(HiSeq/MiSeq)进行测序,为基因组大片断候选基因序列的产出提供了高质量、高通量、低成本的基础,能够确保了测序的真实性和序列的高度精确性以及数据的丰富性,很好的解决了同聚物和重复序列等问题,因此受到了国内外越来越多研究者的青睐。因此本研究通过该方法从分子方面研究白茶芽和叶的代谢途径差异,能够深层地探究茶叶独特的内含物质形成机理。
3.2 茶树富含茶多酚类和多糖类物质,容易与核酸形成不可逆结合,从而导致RNA降解,因此在研究过程中能够采取适当的方法提取RNA为能够达到建库、组装要求的重要前提。本研究的样品在取材后迅速置于液氮中速冻,然后移至-80 ℃冰箱保存,RNA提取时,样品在液氮条件下充分研磨,在液氮刚刚挥发完时,将样品迅速转移至含裂解液的容器中,立即混匀匀浆,最后用TRIzol法提取,使得RNA降解较少,为实验顺利进行提供了可靠地基础。测序结果得到Q30>86,100568条序列,N50(bp)为1904等测序结果表明测序质量相对较好,能够满足转录分析要求。利用NCBI中数据库共注释了unigene数量为100568,NR(NCBI蛋白质非冗余数据库)unigene数量为32530,占比最多为32.35%,KOG(真核生物直系同源基因数据库)unigene数量为30087,占比29.82%,GO(基因本体数据库)unigene数量为 29010,占比28.84%,Swiss-Prot(经实验验证的蛋白序列数据库)unigene数量为24553,占比24.41%,Pfam(蛋白家族数据库)unigene数量为18576,占比18.47%,因此可以看出还有很多基因并没有被注释到相对应的功能中,这种现象在大部分转录组结果中都有出现,这可能与 unigene片段太短有关,数据库基因注释信息缺乏、白茶芽叶中新基因的存在等因素都有关系。
3.3 从样品间基因表达水平相关性统计分析结果看出,同一组织相关系数比较高,不同组织相关性系数不高说明样品选取是有意义的,为之后的基因功能和通路分析提供了保障。通过KEGG通路分析结果可以看出共有16条上调基因调节与类黄酮合成途径有关,13条上调基因调节谷胱甘肽代谢有关,5条下调基因调节光合代谢有关。相同的加工工艺条件下,不同季节茶树鲜叶茶叶品质上有很大的差别,主要是因为内含成分的差别,因此在不同季节会对应的差异基因有所影响。除此之外,环境因素,病虫害等因素都会影响差异基因变化。所以,在之后的研究中将会进一步对茶树相关的代谢产物进行深入的探究。
[1]陈椽.制茶技术理论[M].上海:上海科学技术出版社,1983 CHEN Chuan. Technical theory of tea processing [M].Shanghai: Shanghai Science and Technology Press, 1983
[2]张天福.福建白茶的调查研究[J].茶叶通讯,1963,2(1):43-50 ZHANG Tian-fu. Investigation on white tea distribute in Fujian province [J]. Tea Communication, 1963, 2(1): 43-50
[3]Sharangi A B. Medicinal and therapeutic potentialities of tea(Camellia sinensisL.)-A review [J]. Food Research International, 2009, 42(5): 529-535
[4]宛晓春.茶叶生物化学[M].北京:中国农业出版社,2003 WAN Xiao-chun. Tea biochemistry [M]. Beijing: China Agriculture Press, 2003
[5]Ferrara L, Montesano D, Senatore A. The distribution of minerals and flavonoids in the tea plant (Camellia sinensis)[J]. Il farmaco, 2001, 56(5): 397-401
[6]Manir M M, Kim J K, Lee B G, et al. Tea catechins and flavonoids from the leaves ofCamellia sinensisinhibit yeast alcohol dehydrogenase [J]. Bioorganic & Medicinal Chemistry, 2012, 20(7): 2376-2381
[7]Sekine T, Arita J, Yamaguchi A, et al. Two flavonol glycosides from seeds ofCamellia sinensis[J].Phytochemistry, 1991, 30(3): 991-995
[8]Lakenbrink C, My Loc Lam T, Engelhardt U H, et al. New flavonol triglycosides from tea (Camellia sinensis) [J].Natural Product Letters, 2000, 14(4): 233-238
[9]Morikawa T, Ninomiya K, Miyake S, et al. Flavonol glycosides with lipid accumulation inhibitory activity and simultaneous quantitative analysis of 15 polyphenols and caffeine in the flower buds ofCamellia sinensisfrom different regions by LCMS [J]. Food Chemistry, 2013, 140(1):353-360
[10]Wu Y, Jiang X, Zhang S, et al. Quantification of flavonol glycosides inCamellia sinensisby MRM mode of UPLC-QQQ-MS/MS [J]. Journal of Chromatography B,2016, 1017: 10-17
[11]Juneja L R, Chu D C, Okubo T, et al. L-theanine-a unique amino acid of green tea and its relaxation effect in humans [J].Trends in Food Science & Technology, 1999, 10(6): 199-204[12]Kakuda T. Neuroprotective effects of the green tea components theanine and catechins [J]. Biological and Pharmaceutical Bulletin, 2002, 25(12): 1513-1518
[13]Zheng G, Sayama K, Okubo T, et al. Anti-obesity effects of three major components of green tea, catechins, caffeine and theanine, in mice [J]. In Vivo, 2004, 18(1): 55-62
[14]Yokogoshi H, Kato Y, Sagesaka Y M, et al. Reduction effect of theanine on blood pressure and brain 5-hydroxyindoles in spontaneously hypertensive rats [J]. Bioscience,Biotechnology, and Biochemistry, 1995, 59(4): 615-618
[15]Nathan P J, Lu K, Gray M, et al. The neuropharmacology of L-theanine (N-ethyl-L-glutamine) a possible neuroprotective and cognitive enhancing agent [J]. Journal of Herbal Pharmacotherapy, 2006, 6(2): 21-30
[16]Liang Y R, Liu C, Xiang L P, et al. Health benefits of theanine in green tea: A review [J]. Tropical Journal of Pharmaceutical Research, 2015, 14(10): 1943-1949
[17]文欢,张大燕,彭成,等.天麻苯丙烷代谢途径的转录组学分析[J].中药材,2017,40(4):789-796 WEN Huan, ZHANG Da-yan, PENG Cheng, et al.Transcriptional analysis of phenylpropanoid metabolic pathway inGastrodia elata[J]. Journal of Chinese Medicinal Materials, 2017, 40(4): 789-796
[18]徐振波,侯玉超,刘君彦,等.基因组 De novo组装结合比较RNA-Seq策略在VBNC状态乳杆菌转录组学研究中的应用[J].现代食品科技,2016,32(7):124-129 XU Zhen-bo, HOU Yu-chao, LIU Jun-yan, et al. Application of genome de novo assembly and reference-based transcriptome assembly strategy for studying lactobacilli in the VBNC state [J]. Modern Food Science and Technology,2016, 32(7): 124-129
[19]周子维,李磊磊,孙云.白茶加工工艺及其新品种适制性研究进展[J].中国茶叶加工,2016,26(2):64-68 ZHOU Zi-wei, LI Lei-lei, SUN Yun. Research advance on processing craft and productive character of new tea variaties of white tea [J]. China Tea Processing, 2016, 26(2): 64-68
[20]崔宏春,余继忠,周铁峰,等.白茶主要生化成分比较及药理功效研究进展[J].食品工业科技,2011,32(4):405-408 CUI Hong-chun, YU Ji-zhong, ZHOU Tie-feng, et al.Research progress in main biochemical composition comparasion and pharmacological efficacy of white tea [J].Science and Technology of Food Industry, 2011, 32(4): 405-408
[21]Rio D C, Ares M, Hannon G J, et al. Purification of RNA using TRIzol (TRI reagent) [J]. Cold Spring Harbor Protocols,2010, 2010(6)
[22]Ashburner M, Ball C A, Blake J A, et al. Gene Ontology: tool for the unification of biology [J]. Nature Genetics, 2000, 25(1): 25
[23]Grabherr M G, Haas B J, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome [J]. Nature Biotechnology, 2011, 29(7):644-652
[24]Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences [J].Bioinformatics, 2006, 22(13): 1658-1659
[25]Li B, Dewey C N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome [J].BMC Bioinformatics, 2011, 12(1): 323
[26]Langmead B, Salzberg S L. Fast gapped-read alignment with Bowtie 2 [J]. Nature Methods, 2012, 9(4): 357-359
[27]Mortazavi A, Williams B A, Mccue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq [J].Nature Methods, 2008, 5(7): 621-628
[28]Robinson M D, Mccarthy D J, Smyth G K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data [J]. Bioinformatics, 2010, 26(1):139-140
