气相色谱法测定能力验证样品果蔬汁中毒死蜱含量的不确定度评估
2018-06-07
(大同出入境检验检疫局,大同 037006)
1 前言
测量不确定度是表征合理地赋予被测量量值的分散性与测量结果相联系的参数。一个完整的测量结果除了应给出被测量的最佳估计值之外,还应包括测量不确定度。根据《测试和校准实验室能力的通用要求》(ISO/IEC17025),测量不确定度分析成为近年来计量认证和国家实验室认可评定的重点内容之一。毒死蜱是广泛使用的有机磷类农药,气相色谱法和气相色谱串联质谱法已广泛的用于其定量检测。本文根据国家质量技术监督局发布的JJF1059-2012计量技术规范《测量不确定度评定与表示》,并参照其他文献,对气相色谱法测定果蔬汁中低浓度毒死蜱含量的不确定度进行分析和评定,考察了其不确定度的主要来源,为实验过程的控制和检测结果的评估提供了科学依据。
2 材料与方法
2.1 材料
2.1.1 主要仪器
GC 2010气相色谱仪,日本岛津公司,配有FPD检测器;LE244S型电子天平,赛多利斯公司,感量为0.1mg;10mL、20mL移液管;刻度范围为100~1000μL的移液器;2mL容量瓶;Allegra 64R台式高速冷冻离心机,美国贝克曼库尔特公司。
2.1.2 试剂
毒死蜱标准品:编号为GBW(E)081337,浓度为(1000±7)mg/L,农业部环境保护科研监测所;乙腈、丙酮:色谱纯,天津科密欧化学试剂有限公司。
2.1.3 试验样品
果蔬汁中毒死蜱含量分析样品为能力验证样品,样品编号ACAS-PT244-012,果蔬汁中杀虫剂类农药残留的检测能力验证,组织单位为中国检验检疫科学研究院测试评价中心。
2.2 方法
2.2.1 标准溶液配制
(1)将1000mg/L的毒死蜱标准溶液用丙酮逐级稀释为1.0mg/L和0.1mg/L的毒死蜱标准溶液;
(2)移取1.0mg/L毒死蜱标准溶液125 μL,用丙酮定容至1000μL,稀释成浓度为0.125mg/L的标准溶液;
(3)移取0.1mg/L毒死蜱标准溶液0μL、125μL、250μL、500 μL,用丙酮定容至1000μL,稀释成浓度分别为0、0.0125、0.025、0.05 mg/L的标准溶液。
最终形成0、0.0125、0.025、0.05、0.125mg/L的系列标准溶液。
2.2.2 样品处理
试验前将样品取出解冻,振摇混匀。称取10g样品于离心管中,加入5g氯化钠,用移液管加入20mL乙腈,涡旋5分钟,超声提取30分钟(超声过程保持冰浴)。10000r/min离心3分钟,静置2分钟,从离心管中用移液管吸取10mL上层乙腈溶液,放入鸡形瓶中,置旋转蒸发仪上40℃蒸发近干。冷却后加入1mL丙酮,涡旋混合器上混匀,用0.5mL丙酮洗鸡形瓶两次,涡旋混合器上混匀,转移至2mL容量瓶中,定容至2mL,混匀。过0.22μm有机滤膜,待测[1]。
2.2.3 色谱条件
色谱柱rxt-5型毛细管柱;进样口温度220℃,进样量1μL;柱温程序:50℃(保持2分钟)→150℃(升温速率30℃/分钟,保持2分钟)→210℃(升温速率20℃/分钟,保持7分钟)→260℃(升温速率5℃/分钟,保持15分钟);FPD检测器温度280℃。
3 数学模型和不确定度来源分析
3.1 根据样品前处理过程,果蔬汁中毒死蜱含量按式(1)计算:
(1)
式中:X—样品中毒死蜱的含量,mg/kg;
c—上机试液中毒死蜱的含量,mg/L;
V—上机溶液定容体积,mL;
20—加入提取液乙腈的体积,mL;
m—样品称样量,g;
10—从提取液中分取进行浓缩的溶液体积,mL。
3.2 不确定度来源分析
从测量过程和数学模型分析,果蔬汁中毒死蜱含量不确定度主要来源于标准溶液、仪器的响应值、测量和样品处理操作过程的差异[2],每一种来源又分别受不同因素的影响。其不确定度来源可归纳为A类和B类不确定度。
(1)A类不确定度是由测量重复性引入的不确定度。
(2)B类不确定度主要包括以下几个方面:①标准物质引入的不确定度;②仪器测定引入的不确定度;③样品称量引入的不确定度;④待测样品溶液定容引入的不确定度;⑤前处理液体移取过程引入的不确定度;⑥回收率引入的不确定度[3]。
3.3 不确定度的评定
3.3.1 测量重复性引入的不确定度
在重复性条件下,对本实验所用果蔬汁样品重复测定12次,测定结果见表1。
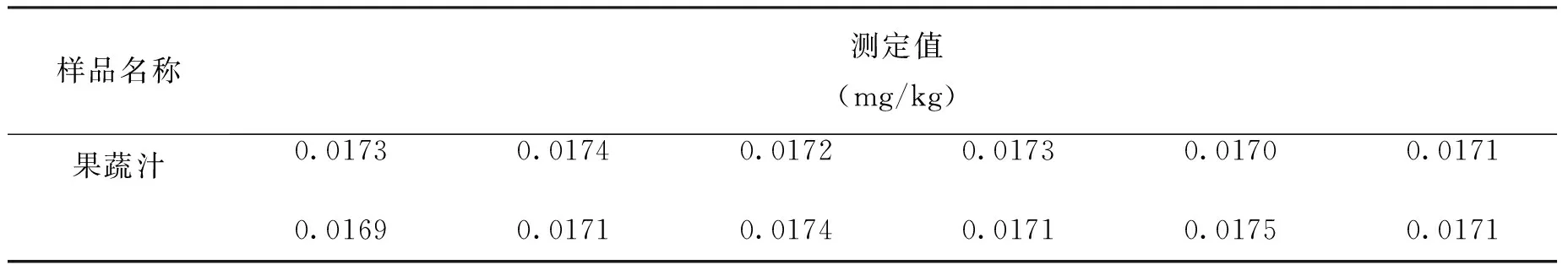
表1 样品重复测定结果
测量重复性的标准不确定度为:
测量重复性引入的相对标准不确定度为:
3.3.2 毒死蜱标准物质引入的不确定度
(1)标准品引入的不确定度
编号为GBW(E)081337毒死蜱标准品浓度为(1000±7)mg/L,相对标准不确定度为urel(P)=7/1000=7×10-3。
(2)标准溶液配制过程中移液器引入的不确定度。
配制过程中的不确定度主要由移液器的校准、量取的变动性、环境因素产生。试验过程控制在20℃的环境温度下进行,温度的影响可以忽略;量取的变动性在方法的重复性中已做评定,不再重复;故移液器的校准为主要的不确定度来源。刻度范围为100~1000μL的移液器的最大允许误差为:±10μL,按均匀分布处理[4],其标准不确定度及相对标准不确定度分别为:
(3)毒死蜱标准物质的相对标准不确定度
由标准物质本身、校准溶液配制过程中移液器校准的不确定度分量合成标准溶液的相对标准不确定度:
3.3.3 仪器测定引入的不确定度
(1)仪器数据处理系统引入的不确定度
根据仪器说明书和积分仪的一般性能指标分析,目前用于气相色谱仪峰面积积分处理的最大误差为0.2%~1%,取1%,按均匀性分布考虑,则相对不确定度为:
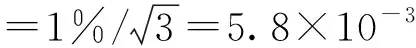
(2)峰面积重复测量产生的不确定度
通过重复进样进行评定,它包含试样进样量的不确定度,该不确定度属A类,数据见表2。

表2 重复进样峰面积测试结果
当n=3,极差系数C=1.69,标准不确定度:u(A)=R/C=1.2×103
相对标准不确定度:urel(A)=u(A)/A=0.0063
(3)仪器测定引入的不确定度的合成
合成毒死蜱试样处理液峰面积测量的相对标准不确定度为:
3.3.4 待测样品称量引入的不确定度
用万分之一天平称取样品10.0000g,天平的最大允许误差为±0.0010g,按均匀分布处理,其标准不确定度及相对标准不确定度分布为:
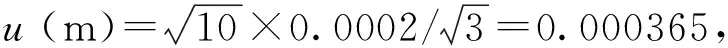
urel(m)=0.000365/10.0000=3.6×10-5
3.3.5 待测样品溶液定容引入的不确定度
A级2mL容量瓶最大允许误差为±0.015mL,按均匀分布处理,其标准不确定度及相对标准不确定度分别为:
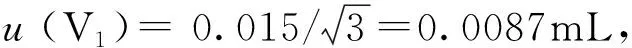
urel(V1)=0.0087/2=4.3×10-3
3.3.6 前处理液体移取过程引入的不确定度
A级10mL单标线移液管、20mL单标线移液管的最大允许误差为±0.020mL、±0.030mL,按均匀分布处理[5],其标准不确定度及相对标准不确定度分别为:
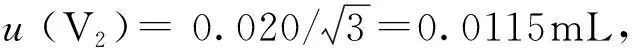
urel(V2)=0.0115/10=1.2×10-3
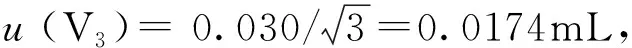
urel(V3)=0.0174/20=8.7×10-4
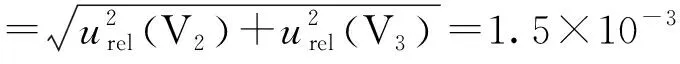
3.4 回收率引入的不确定度
由于样品前处理不完全或处理过程中导致毒死蜱的损失或污染等,使用同一方法做加标回收试验,回收率结果见表3。
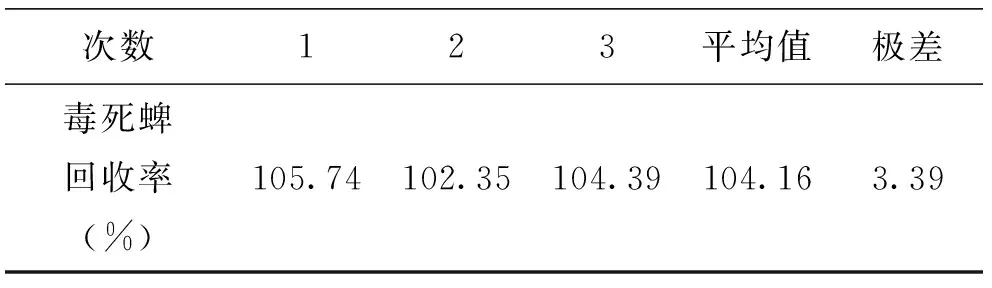
表3 毒死蜱加标回收率
当n=3,极差系数C=1.69,标准不确定度:u(Rec)=R/C=2.006%
相对标准不确定度:urel(Rec)=u(Rec)/Rec=0.0193
3.5 合成标准不确定度
由于上述数学模型仅涉及积和商的数学关系,其合成标准不确定度可简化计算如下:
=0.025X
表1中CX=0.0172mg/kg,则其合成标准不确定度为
ucrel(X) =0.025×0.0172 mg/kg
=0.0004mg/kg
3.6 扩展不确定度
扩展标准不确定度可由合成标准不确定度乘以包含因子,取k=2(置信区间95%),得扩展不确定:U=ucrel(X)×k=0.0008mg/kg
3.7 测定不确定度的报告
样品中毒死蜱含量为:(0.0172±0.0008)mg/kg,k=2。
4 结论
对果蔬汁中毒死蜱含量测量过程引入的不确定度分量进行评估,最后计算相对合成不确定度,求出毒死蜱含量的扩展不确定度。对各分量的不确定度比较发现,样品处理回收率、仪器、标准物质、重复性实验对测定结果不确定度的影响大。因此在试验中,可以从提高仪器的精密性、操作过程的一致性等方面提高测量结果的准确性[6]。
[1] NY/T 761-2008 蔬菜和水果中有机磷、有机氯、拟除虫菊酯和氨基甲酸酯类农药多残留的测定.
[2] 王秋艳.高效液相色谱法测定果汁饮料中苯甲酸含量的不确定度评定[J],检验检疫学刊,2015,25(2),33-35.
[3] 顾宗理,分光光度法测定水质总氰化物含量的不确定度评定[J],化学世界,2011,04(7),397-400.
[4] JJG 646-2006移液器检定规程[S].北京:中国计量出版社,2006.
[5] JJG 196-2006 常用玻璃仪器检定规程[S].北京:中国计量出版社,2006.
[6] 吴俐,气相色谱法测定稻米中毒死蜱的不确定度[J],中国稻米,2017,23(1),54-56.
