基于SNP位点的身高预测模型的评估
2018-06-04焦会永孙亚男景晓溪李彩霞黄艳梅赵雯婷
焦会永 ,孙亚男 ,景晓溪 ,刘 京 ,江 丽 ,李彩霞 ,叶 健 ,刘 凡 ,黄艳梅 ,赵雯婷
(1.新乡医学院法医学系,河南 新乡 453003;2.公安部物证鉴定中心 北京市现场物证检验工程技术研究中心 现场物证溯源技术国家工程实验室 法医遗传学公安部重点实验室,北京 100038;3.济宁医学院法医学与医学检验学院,山东 济宁 272067;4.中国科学院北京基因组研究所,北京 100029)
通过DNA进行人类身高、外貌等表型特征的预测,是目前法医DNA领域的研究热点之一[1-3]。身高特征由于比较容易识别、可以量化、成年后相对稳定,成为描述和识别个体特征的重要指标,对于案件侦查具有重要的意义。研究[4]表明,身高是典型的多基因遗传性状,受遗传、环境等因素影响。其中,遗传因素对身高的影响较环境因素更加明显,占80%左右[5]。目前国内外研究报道了大量身高相关基因、SNP位点及稀有变异等遗传信息[6]。
利用法医人类学方法对身高进行预测的研究取得了一定的进展[7-8],然而可以应用于实际办案的身高预测技术方法仍然较少。在法医DNA领域,目前构建的身高预测模型主要是针对欧洲极高身高个体的定性研究,使用的主要方法有通过利用分类与回归树(classification and regression tree,CART)法、支持向量机(support vector machine,SVM)法及加权等位基因求和(weighted allele sums,WAS)等模型来预测身高[9]。AULCHENKO等[10]首次报道了采用WAS模型针对54个SNP位点进行荷兰人群中5%的极高身高个体的定性预测研究,其预测准确性的曲线下面积(area under curve,AUC)值为 0.65,比随机预测的 0.5稍高。LIU等[9]基于欧源样本的研究建立了180个SNP位点的WAS预测模型,极高身高个体的定性预测准确性提升到了0.75。2014年,WOOD等[11]以包含253 288个欧洲个体的79个研究项目为基础,通过Meta分析得到了697个与身高显著关联的SNP位点,是目前国际上身高研究领域影响力较大的一项研究成果。目前,针对亚洲人群的身高模型研究还较少。本研究拟依据WOOD等[11]研究中相关SNP位点构建身高预测模型,评估其在中国山东汉族男性群体中的预测能力。
1 对象与方法
1.1 研究对象
样本采集前对志愿者进行问卷调查,主要调查包括三代以内亲属的民族、年龄及是否患有影响身高的相关疾病等问题,筛选出符合标准的志愿者进行样本采集。纳入标准:(1)三代以内均为汉族且无异族通婚史;(2)年龄为 20~50 岁的男性;(3)身高≥180cm(高身高)或≤165cm(低身高)。排除标准:(1)曾接受过激素治疗;(2)有先天性发育异常或后天器质性畸形;(3)患有甲状腺疾病、垂体疾病及肿瘤等;(4)患有身高缺陷性、家族遗传性、精神性、影响骨代谢及其他影响生长发育的急慢性疾病。本研究共采集了59例山东汉族男性无关个体(表1)的血液样本,分别用于全基因组关联分析和二代测序,所有志愿者均签署知情同意书。

表1 研究对象一般资料
身高由经过专业培训的人员进行测量,采用统一的测量工具及测量方法。测量方法:被测量者呈立正姿势,赤脚站立于身高测量计的底板上,足跟并拢,足尖分开约呈60°,上肢自然下垂,肩甲、骶骨及脚跟紧靠身高测量计的立柱,双眼平视前方;测量者站在被测量者一侧,调整身高测量计平板至适宜位置,记录身高数据,精确至1cm。
1.2 方法
1.2.1 DNA提取与定量
使用 QIAamp DNA Blood Midi试剂盒(德国QIAGEN公司)提取所有样本的全基因组DNA,采用NanoDrop 2000分光光度计(美国Thermo Scientific公司)检测DNA浓度。
1.2.2 SNP分型检测
全基因组关联分析(Genome-wide association study,GWAS)研究:采用 Affymetrix SNP Array 6.0芯片(美国Thermo Fisher Scientific公司)对42例山东汉族男性样本进行基因分型。用Sty或NSP酶对250ng DNA样本进行消化,对消化后的小片段DNA进行连接反应,然后用通用引物进行PCR扩增。扩增反应完成后,对产物进行片段化处理并用生物素对片段进行标记,再与芯片进行杂交。经过16~18h的杂交反应,采用GeneChipTMScanner 3000 7G(美国AB公司)检测杂交信号荧光强度并读取数据。
二代测序:采用HiSeq 4000测序平台(美国Illumina公司)对17例山东汉族男性样本进行全基因组测序,经过BWA软件(美国Illumina公司)[12]将测序短序列与参考基因组进行比对,确定短序列在基因组上的位置,使用SAMtools软件(美国Illumina公司)[13]将这些短序列按一定顺序排列并进行数据转换,通过Picard软件(美国博德研究所)进一步去除测序过程中产生的冗余信息及噪声,使用GATK软件(美国博德研究所)寻找SNP位点,最后使用ANNOVAR软件[14-15]对SNP位点进行功能注释。17例样本的DNA测序检测委托北京市理化分析测试中心及中国科学院北京基因组研究所进行。
1.2.3 数据处理及统计学分析
针对芯片检测获得的SNP分型结果采用GCTA软件[16]得到样本之间的遗传相关矩阵(genetic relationship matrix,GRM),未检测到有强亲缘关系的样本。对GRM进行奇异值分解(singular value decomposition,SVD)得到主要的特征向量(eigenvector),对主要的特征向量进行k-means聚类,从而进行人群分层评估,并将主要的特征向量在GWAS中作为协变量以校正可能存在的人群亚分层混淆因素。GWAS分析中用PLINK v1.9软件(美国博德研究所)[17]的Logistic回归检测基因型-表型相关性,因变量为logit(P),其中P为低身高的概率,低身高设为1,高身高设为0;SNP分型及PLINK v1.9软件[17]分析得到的三个主成分(C1、C2及C3)为自变量,自变量SNP基因型以效应频率等位基因数目赋值,即非效应等位基因纯合型赋值为0、杂合型赋值为1,效应等位基因纯合型赋值为2,挖掘与表型显著相关的SNP位点(P≤0.05认为与身高潜在关联,P≤5×10-8认为与身高达到全基因组水平显著关联[11])。使用Mathematical软件[18]对GWAS数据进行基因填补[5,19-20],从得到的相应SNP分型数据中筛选出与参考文献[11]共有的SNP位点信息。
17例样本的测序数据,使用PLINK v1.9软件[17]进行主成分分析以去除人群分层,提取SNP位点分型信息,与上述GWAS分型结果进行合并分析。由于样本量较少,这17例样本未进行GWAS分析。
采用R v3.2.2软件绘制GWAS分析Q-Q plot图及曼哈顿图,通过Q-Q plot图直观显示观测值与预测值之间的差异,判断图中的SNP位点分布是否合理,以排除潜在的偏差SNP位点,如分型错误;通过曼哈顿图直观显示SNP位点与身高的关联性。
1.3 身高预测模型建立
首先根据参考文献[11]中报道的697个SNP位点效应等位基因的Beta值(其值大小和正负与该等位基因对身高的作用呈正相关),获得每个样本的WAS值,即:
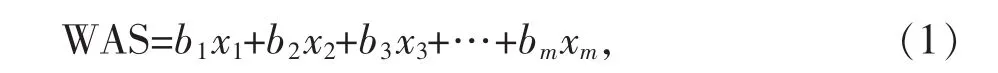
其中,bm为第m个SNP位点的效应等位基因的Beta值,xm为每个样本第m个SNP位点的效应等位基因数目。继而依照以二进制身高为因变量(y)的Logistic回归模型:

计算每个样本为高身高或低身高的概率,即:

然后参考LIU等[9]的研究方法,使用SPSS 19.0软件根据样本数据绘制受试者工作特征(receiver operating characteristic,ROC)曲线并计算其曲线下面积(area under curve,AUC),采用 AUC 值评价预测模型的准确性,AUC值分布为0.5~1.0,0.5表示没有预测能力,数值越接近1.0表示模型的预测能力越准确[21]。
将芯片筛选结果与二代测序提取结果合并,筛选出共有的SNP位点,用于身高预测模型准确性评价。针对上述样本数据,用Excel软件(美国Microsoft公司)绘制散点图,使用SPSS 19.0软件(美国IBM公司)对模型所用SNP位点的Beta值在本研究与参考文献[9]中的差异进行卡方检验,并使用R v3.2.2软件绘制高身高、低身高分布密度曲线。
2 结 果
2.1 GWAS结果
采用Affymetrix SNP Array 6.0芯片进行检测的42例样本共检测了909622个SNP位点,使用PLINK v1.9软件对检测的全部SNP位点进行质量控制:(1)剔除分型成功率小于90%的样本2个;(2)剔除分型成功率小于90%的SNP位点90 039个;(3)剔除效应等位基因最小等位基因频率(minor allele frequency,MAF)小于0.01的SNP位点161595个。最终共得到40例样本的657988个SNP位点用于身高关联分析。
二代测序17例样本的质量控制:(1)剔除分型成功率小于90%的染色体位点5489036个;(2)剔除较小等位基因频率小于0.05的染色体位点1047 604个。最终共获得17例样本的3123513个SNP位点。
通过PLINK v1.9软件中的主成分分析(principal component analysis,PCA)模块将个体的高维度(维度等于该个体检测的SNP位点数目)全基因组SNP位点分型数据降到低维度(维度不大于个体数目)。分析得到的前三个主成分(C1、C2及C3)累积贡献率〉85%,故以这三个主成分作为协变量加入到关联分析模型中,进一步校正可能的人群分层[22],17例样本和40例样本的分析均未发现人群分层,Q-Q plot图可见样本存在较高的偏度和峰度(图1A)。通过GWAS样本分析,绘制曼哈顿图显示无达到全基因组水平显著关联 SNP位点(P≤5×10-8),未发现与身高显著相关的SNP 位点(图 1B)。
2.2 用WAS法构建中国汉族身高预测模型
40例样本的分型结果与参考文献[11]中697个SNP位点的共有位点为186个,经基因填补[23]后达到了612个。对二代测序的17例样本分型结果进行提取,得到595个SNP位点。两者统一整合后筛选出547个共有的SNP位点,并将效应等位基因(频率较小的等位基因)与参考文献[11]进行比对,根据效应等位基因是否一致调整Beta值方向,进而获得适用于构建本研究身高预测模型的Beta值,构建WAS预测模型。利用身高预测模型绘制ROC曲线并计算其AUC值为0.67(95%置信区间为0.53~0.90)。根据1.3节的公式获得WAS值绘制密度曲线(图2),提示此模型对高身高(30例)及低身高(27例)的预测有一定区分度。

图1 身高关联研究Logistic回归分析结果
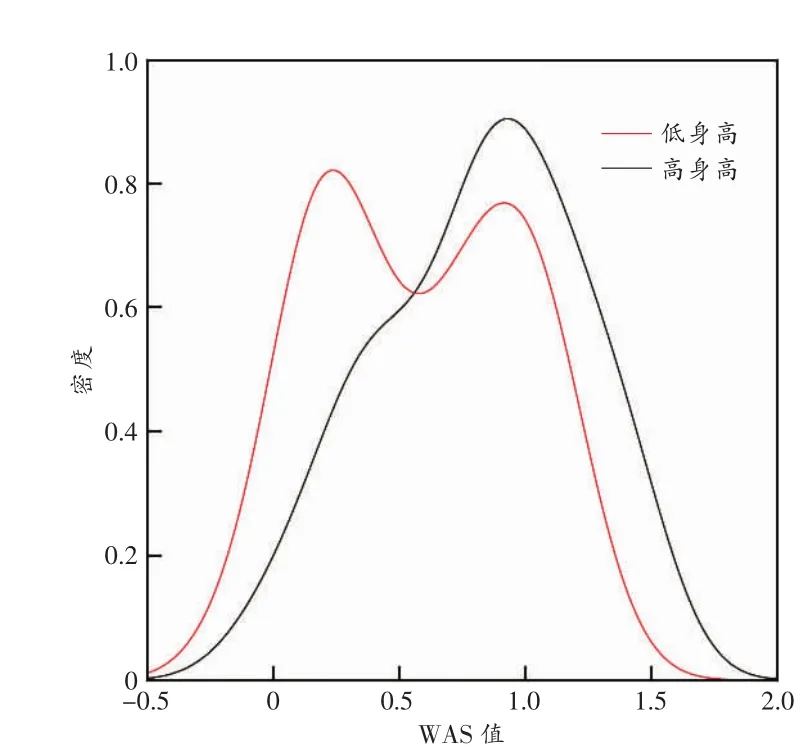
图2 57例样本身高的WAS密度曲线
身高相关SNP位点对人类身高的影响具有人群差异。Beta值方向分布散点图(以两组Beta值交点为原点,处在第一、三象限的Beta值方向相同,第二、四象限的Beta值方向相反)显示,本研究与WOOD等[11]研究人群之间Beta值方向分布有较大差异(图3),提示身高相关SNP位点存在种族特异性。针对欧洲及中国汉族547个SNP位点对应Beta值在高身高及低身高水平的分布(表3)进行卡方检验,χ2=6.934,双侧精确P=0.008,单侧精确P=0.040,两者Beta值之间差异具有统计学意义,进一步提示种族特异性的存在。
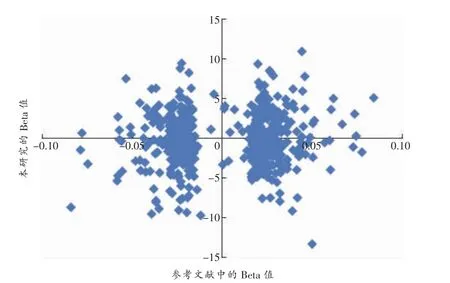
图3 547个SNP位点对应Beta值在本研究及参考文献[11]中的分布
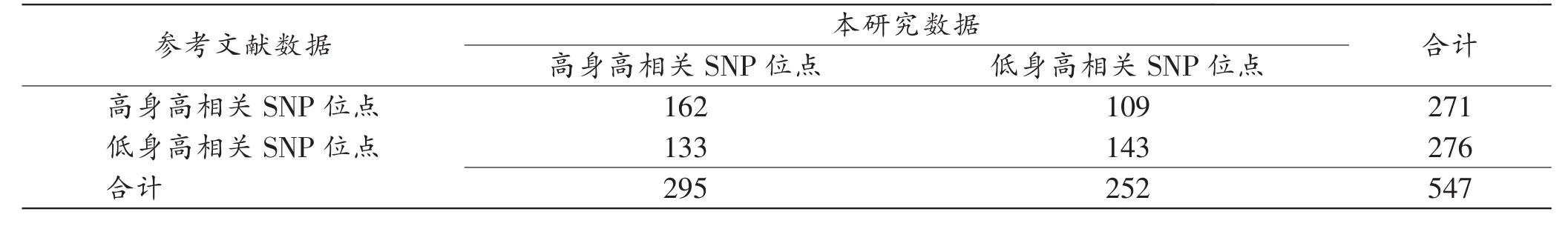
表3 547个SNP位点分布情况 (个)
3 讨 论
身高是由遗传和环境因素共同影响的复杂多基因性状,随着新一代测序技术及GWAS的推广,大量身高相关SNP位点被发现。近几年,国外不少研究开始尝试基于身高相关SNP位点来预测人类身高[9]。对人类身高的预测研究在临床诊断、法庭科学等多个领域都有潜在的应用价值。在临床方面,如果能从DNA水平预测未来身高发育方面的异常,有助于医生尽早诊断先天性巨人症及生长限制类疾病并及时进行干预[9]。在法医学实践方面,通过DNA数据预测个体的身高信息,可以为案件侦查提供线索。
由于国内尚未发现足够数量的中国汉族人群身高显著相关SNP位点,因此无法直接构建用于预测中国汉族人群身高的预测模型。本研究选取了当前国际上在身高预测方面效果较好的欧洲高加索人群身高显著相关SNP位点,构建预测模型,用于评估其预测中国汉族人群身高的准确性。评价标准即为ROC曲线的AUC。ROC曲线源于信号探测理论,最初用于描述信号与噪声之间的关系,以评价雷达性能[24],随着ROC分析应用于医学诊断,日益受到重视,到目前已经发展成为一种分析方法。ROC曲线采用二分法进行分析,以真阳性率(灵敏度)为纵坐标、假阳性率(特异性)为横坐标,AUC越大,判断准确性就越大[25]。目前,国内外人类身高预测模型较少,WAS即为其中之一且预测准确性较高[9]。AULCHENKO等[10]首次将ROC曲线应用于身高预测,根据每例样本的WAS值绘制ROC曲线,构建身高预测模型,在仅有54个身高相关SNP位点的情况下,AUC值达到了0.65。随后,LIU等[9]根据180个身高相关SNP位点计算每例样本WAS值并进行了ROC分析,其AUC值达到了0.75(95%置信区间为 0.72~0.79)。
目前,中国人群身高相关遗传位点研究还处于起步阶段,需要进行人群遗传多态性分析、人群身高模型构建、极端身高个体定性、个体身高预测等多个阶段的研究,实现对实际案件生物检材来源人身高的精细推测还有很长的路要走。本研究初步构建了可用于中国汉族人群身高预测的计算模型并进行效果评估,借鉴AULCHENKO等[10]研究方法来评估此模型预测中国汉族人群身高的准确性,为后续研究提供了数据支撑。本研究首先应用Affymetrix Array SNP 6.0芯片对40例中国汉族男性样本进行GWAS研究,仅发现了与身高潜在相关的SNP位点,但在全基因组水平未发现与身高显著相关的SNP位点。可能由以下原因导致:(1)样本量过少;(2)大部分与身高相关的SNP位点效应较小,很难被检测到。本研究采用WAS分析法获得的AUC值为0.67(95%置信区间为0.53~0.90)。与AULCHENKO等[10]研究相比,本研究在使用的SNP位点数量大大提升的条件下,预测准确性稍高,但仍低于LIU等[9]在欧洲人群中的研究。另外,本研究身高预测模型所用的547个身高相关SNP位点是基于欧洲人群研究得出的,而卡方检验结果提示身高相关SNP位点存在种族差异性[26],推测本研究预测模型AUC值比LIU等[9]低的主要原因,是因为在欧洲人群发现的身高相关SNP对中国汉族人群并不完全适用。
在后续的研究工作中,需要关注身高遗传学研究的最新进展,增加用于筛选验证的相关SNP位点;加大人群验证样本量,不断挖掘适用于中国汉族人群的身高相关SNP位点;改善计算方法体系,继续优化模型算法。
[1]KAYSER M.Forensic DNA Phenotyping:Predicting human appearance from crime scene material for investigative purposes[J].Forensic Sci Int Genet,2015,18:33-48.
[2]TURKHEIMER E.Genetic Prediction[J].Hastings Cent Rep,2015,45(S5):32-38.
[3]WALSH S, LIU F, WOLLSTEIN A, et al.The HIrisPlex system for simultaneous prediction of hair and eye colour from DNA[J].Forensic Sci Int Genet,2013,7(1):98-115.
[4]杨应忠,王亚平,马兰,等.中国汉族高原肺水肿易感基因的全基因组关联研究[J].遗传,2013,35(11):1291-1299.
[5]DU M,AUER P L,JIAO S,et al.Whole-exome imputation of sequence variants identified two novel alleles associated with adult body height in African Americans[J].Hum Mol Genet,2014,23(24):6607-6615.
[6]CHO Y S, GO M J, KIM Y J, et al.A largescale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits[J].Nat Genet,2009,41(5):527-534.
[7]THODBERG H H,JENNI O G,CAFLISCH J, et al.Prediction of adult height based on automated determination of bone age[J].J Clin Endocrinol Metab,2009,94(12):4868-4874.
[8]MARTIN D D, SCHITTENHELM J, THODBERG H H.Validation of adult height prediction based on automated bone age determination in the Paris Longitudinal Study of healthy children[J].Pediatr Radiol,2016,46(2):263-269.
[9]LIU F, HENDRIKS A E,RALF A, et al.Common DNA variants predict tall stature in Europeans[J].Hum Genet,2014,133(5):587-597.
[10]AULCHENKO Y S,STRUCHALIN M V, BELONOGOVA N M,et al.Predicting human height by Victorian and genomic methods[J].Eur J Hum Genet,2009,17(8):1070-1075.
[11]WOOD A R, ESKO T, YANG J, et al.Defining the role of common variation in the genomic and biological architecture of adult human height[J].Nat Genet,2014,46(11):1173-1186.
[12]LI H,DURBIN R.Fast and accurate short read alignment with Burrows-Wheeler transform[J].Bioinformatics,2009,25(14):1754-1760.
[13]LI H.A statistical framework for SNP calling,mutation discovery,association mapping and population genetical parameter estimation from sequencing data[J].Bioinformatics,2011,27(21):2987-2993.
[14]WANG K,LI M,HAKONARSON H.ANNOVAR:functional annotation of genetic variants from high-throughput sequencing data[J].Nucleic Acids Res,2010,38(16):e164.
[15]YANG H,WANG K.Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR[J].Nat Protoc,2015,10(10):1556-1566.
[16]YANG J, LEE S H,GODDARD M E,et al.GCTA:a tool for genome-wide complex trait analysis[J].Am J Hum Genet,2011,88(1):76-82.
[17]CHANG C C,CHOW C C,TELLIER L C,et al.Second-generation PLINK:rising to the challenge of larger and richer datasets[J].Gigascience,2015,4:7.
[18]LIU E Y,LI M,WANG W,et al.MaCH-admix:genotype imputation for admixed populations[J].Genet Epidemiol,2013,37(1):25-37.
[19]ESTRADA K,KRAWCZAK M,SCHREIBER S,et al.A genome-wide association study of northwestern Europeans involves the C-type natriuretic peptide signaling pathway in the etiology of human height variation[J].Hum Mol Genet,2009,18(18):3516-3524.
[20]LI Y,WILLER C,SANNA S,et al.Genotype imputation[J].Annu Rev Genomics Hum Genet,2009,10:387-406.
[21]陈卫中,倪宗瓒,潘晓平,等.用ROC曲线确定最佳临界点和可疑值范围[J].现代预防医学,2005,32(7):729-731.
[22]PRICE A L,PATTERSON N J,PLENGE R M,et al.Principal components analysis corrects for stratification in genome-wide association studies[J].Nat Genet,2006,38(8):904-909.
[23]何桑,丁向东,张勤.基因型填充方法介绍及比较[J].中国畜牧杂志,2013,49(23):95-100.
[24]SWETS J A.Measuring the accuracy of diagnostic systems[J].Science,1988,240(4857):1285-1293.
[25]王敬瀚.ROC曲线在临床医学诊断实验中的应用[J].中华高血压杂志,2008,16(2):175-177.
[26]LEI S F, TAN L J, LIU X G, et al.Genome-wide association study identifies two novel loci containing FLNB and SBF2 genes underlying stature variation[J].Hum Mol Genet,2009,18(9):1661-1669.
