基于[Rh(CO)2I2]-的Rh配合物催化剂的分子设计
2018-06-01吉文欣王殿军楚秀秀
吉文欣,王殿军,楚秀秀
(1.宁夏大学 省部共建煤炭高效利用与绿色化工国家重点实验室, 宁夏 银川 750021;2.宁夏大学 化学化工学院,宁夏 银川 750021)
乙酸是一种重要的基本有机化工产品,作为重要原料和一种优良的有机溶剂广泛应用于化工、轻纺、塑料、医药、橡胶以及染料等行业。我国是乙酸的生产大国,也是乙酸的消耗大国,2014年中国乙酸产能已经达到9.72 Mt/a,其中采用羰基铑碘化合物([Rh(CO)2I2)]-)催化剂为核心技术的甲醇羰化法工艺已经占我国乙酸总产能的85%以上。该催化剂对甲醇羰基化合成乙酸具有较高的催化活性和选择性[1],但其稳定性有待提高,在CO不足时(如在生产后期分离产物闪蒸过程中) Rh(I)易被氧化为Rh(III),进而从体系中沉淀出来,导致催化剂失活;为解决这个问题,通常在反应中加入大量水,这会造成物料浓度下降,产能降低,后期产物分离能耗增加。因此,在保证较高催化活性的同时,如何防止催化剂在后期分离时失活,是该领域的热点问题[2-5]。
催化剂稳定性的提高必须是在保证其活性和选择性的前提下才是有意义的,而盲目的实验往往顾此失彼。利用催化剂的分子设计,通过理论计算确认催化反应机理,得到反应能垒,在分子水平上研究催化剂结构与性能的内在关系,以便合成特定功能的催化剂。图1为[Rh(CO)2I2)]-催化剂失活机理[6]。由图1可见,Rh(I)的失活是因为2分子催化剂反应生成二聚体后Rh(I)氧化变价生成RhI3沉淀,所以提高催化剂的稳定性,必须阻止催化剂的分子间反应形成二聚体。
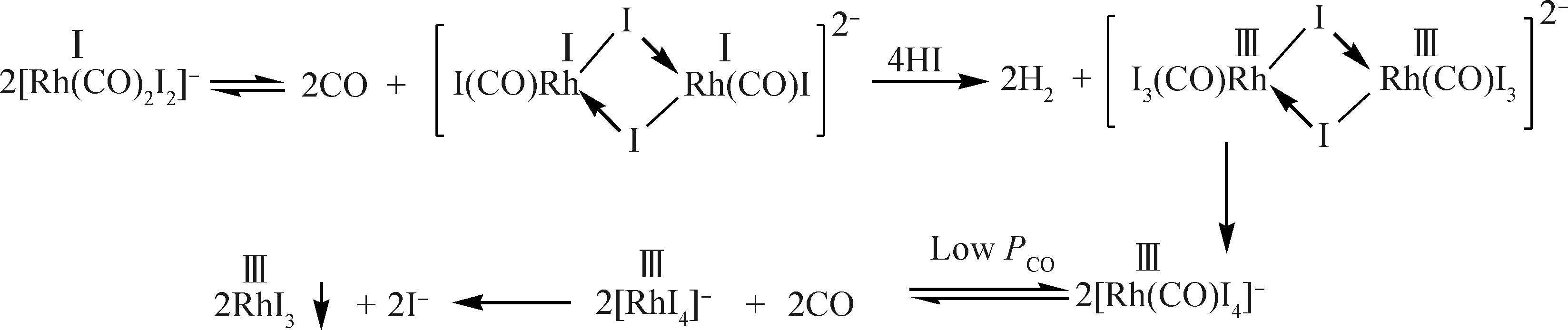
图1 [Rh(CO)2I2)]-催化剂甲醇羰基化反应失活机理[6-8]Fig.1 Diagram of deactivated mechanism for methanol carbonylation over [Rh(CO)2I2)]- catalyst[6-8]Ⅰ—Rh+1; Ⅲ—Rh+3
已有研究表明,含有N、P和O的基团与Rh配位,可以在一定程度上提高[Rh(CO)2I2)]-催化剂的稳定性和活性[9-10]。[Rh(CO)2I2)]-催化甲醇羰基化制备乙酸反应分四步进行:碘甲烷氧化加成反应、羰基重排反应、羰基配位反应、CH3COI还原消除基元反应[11-14]。其中碘甲烷氧化加成反应能垒最高,是整个反应的决速步骤。要增加催化剂活性必须降低碘甲烷氧化加成反应能垒。前期研究[15]表明,含有N、O的基团可以增加中心原子Rh上的富电子性,并促进CH3I的亲核取代反应,降低碘甲烷氧化加成反应能垒,提高催化剂活性。另一方面,采用能与Rh(I)形成稳定化合物的配体,利用其空间位阻效应阻止 [Rh(CO)2I2)]-的分子间反应,防止Rh(I)的变价沉淀,进而提高催化剂稳定性,减少水的添加量,在增加产能的同时降低分离能耗。
1 研究方法
采用有效核势能近似(ECP,effective core potential approximation),MP2(Møller-Plesset theory,2nd order)方法,在SDD基组水平上,以羧基吡啶作为配体,与Rh(I)阳离子形成一系列包含不对称双齿结构的羧基吡啶铑阳离子催化剂:邻-吡啶丙酸铑阳离子([o-Py-C2H4COOH-Rh(CO)2]+)、间-吡啶丙酸铑阳离子([m-Py-C2H4COOH-Rh(CO)2]+)、对-吡啶丙酸铑阳离子([p-Py-C2H4COOH-Rh(CO)2]+)、邻-吡啶丁酸铑阳离子([o-Py-C3H6COOH-Rh(CO)2]+)、间-吡啶丁酸铑阳离子([m-Py-C3H6COOH-Rh(CO)2]+)、对-吡啶丁酸铑阳离子([p-Py-C3H6COOH-Rh(CO)2]+),计算得到了系列催化剂的稳态构型,分析该系列催化剂构型、能量以及中心原子Rh上的电荷和电子密度,对比[Rh(CO)2I2)]-催化剂,通过分析催化剂构型、电荷分布等与活性、稳定性之间的内在关系,探讨了催化剂具有更高反应活性和稳定性的本质原因,设计出了一种具有更高活性和稳定性的催化剂。所有计算采用Gaussain 09量子化学计算软件完成。
2 结果与讨论
2.1 催化剂构型
图2为羧基吡啶铑阳离子系列催化剂构型。由图2可知,在[o-Py-C2H4COOH-Rh(CO)2]+的羧酸碳链上插入1个CH2,就得到了[o-Py-C3H6COOH-Rh(CO)2]+,如果进一步改变羧基在吡啶环上的位置,就会得到位于邻位(o-)、间位(m-)和对位(p-)的同分异构体系列催化剂。计算结果表明,6种催化剂都存在较稳定的构型,Rh—N键长在0.2078~0.2099 nm之间,相比之下,Rh—O键的键长更长(0.2112~0.2196 nm),键能更小,∠N—Rh—O在74.7°~92.7°之间。Rh与配体之间形成了七元环、八元环的构型,分子骨架易发生扭动,这样的结构将有利于协同碘甲烷氧化加成反应中催化剂的结构挠变,降低碘甲烷氧化加成反应能垒。
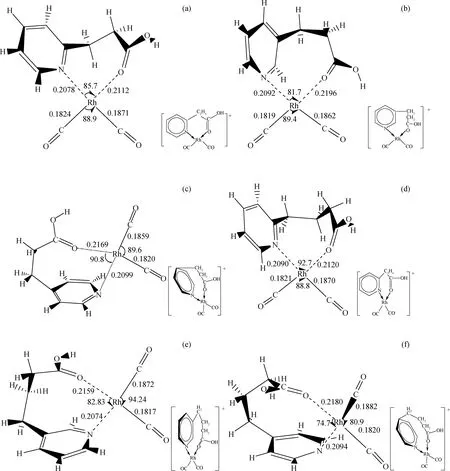
图2 羧基吡啶铑阳离子系列催化剂构型Fig.2 Optimized geometric parameters of carboxylic pyridine Rh(I) catalysts at MP2/SDD level Bond length in nm, and bond angle in degree(a) [o-Py-C2H4COOH-Rh(CO)2]+; (b) [m-Py-C2H4COOH-Rh(CO)2]+; (c) [p-Py-C2H4COOH-Rh(CO)2]+; (d) [o-Py-C3H6COOH-Rh(CO)2]+; (e) [m-Py-C3H6COOH-Rh(CO)2]+; (f) [p-Py-C3H6COOH-Rh(CO)2]+
2.2 催化剂中心原子Rh电荷、电子密度与不对称配位结构
对于以Rh(I)为活性组分的甲醇羰基化催化剂来说,催化反应的决速步骤是CH3I与活性中心Rh的氧化加成反应,这也可以看成是Rh对CH3I的亲核取代反应,若能提高其亲核能力,即可提高催化剂活性,通过增加Rh的富电子性可实现这一目的。在分子设计中,采用含吡啶和羧酸的给电子基团与Rh(I)配位,可以有效增加中心原子Rh的电子密度,即富电子性。表1为[Rh(CO)2I2)]-、羧基吡啶铑阳离子系列催化剂Rh上的电荷数以及电子密度。由表1可见,6种催化剂的Rh都带正电,其中[p-PyC2H4COOHRh(CO)2]+中Rh的Mulliken电荷最低,而[o-PyC3H6COOHRh(CO)2]+中Rh上的电子密度最大,但总体看,Rh上的电荷数和电子密度的区别不大,这说明采用羧基吡啶铑阳离子催化剂中心原子Rh的富电子性相似。
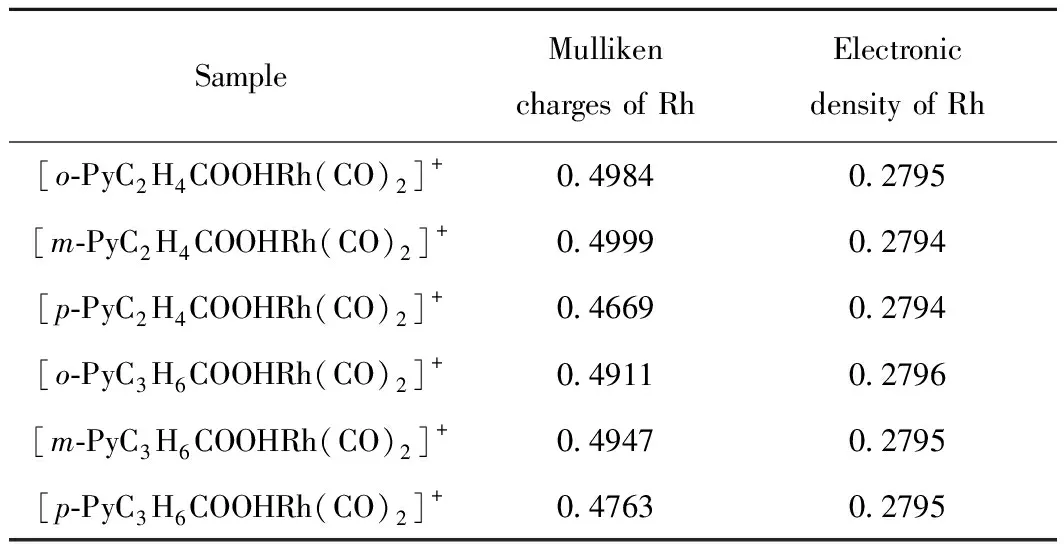
表1 [Rh(CO)2I2)]-、羧基吡啶铑阳离子系列催化剂Rh上的电荷数以及电子密度Table 1 The electric charge and electronic density of Rh in [Rh(CO)2I2)]- and carboxylic pyridine Rh(I) catalysts
图3为[Rh(CO)2I2)]-和[o-PyC2H4COOHRh(CO)2]+与CH3I氧化加成反应过程中各驻点几何结构。采用具有不对称双齿结构的羧基吡啶配体与Rh(I)配位,形成一短一长,一强一弱的Rh—N、Rh—O配位键,在决速步骤CH3I加成反应中(如图3(d)[o-PyC2H4COOHRh(CO)2]+),Rh被1个较强的Rh—N配位键牢牢“抓住”;而Rh—O键,反应前为0.2112 nm,CH3I加成反应开始后,Rh—O键不断拉长,到达过渡态(TS2)时基本断开(0.4576 nm),CH3I加成反应结束后,Rh—O键恢复(0.2130 nm)。可以预见,1个相对较弱的Rh—O键将更有利于协同CH3I加成反应,有效降低反应能垒,增加催化剂活性。表2为[Rh(CO)2I2)]-、羧基吡啶铑阳离子系列催化剂中心原子Rh与周围原子间键鞍点电子密度的Laplacian量▽2ρ(r)。由表2可知,不同配体的催化剂,只有Rh—N、Rh—O 间的键鞍点电子密度的Laplacian量▽2ρ(r)有一定差别,其中[o-PyC2H4COOHRh(CO)2]+的差别最大,说明该催化剂的Rh—N、Rh—O间的强弱差别最大,而它的Rh—O间键鞍点电子密度的Laplacian量▽2ρ(r)在6种催化剂中正值最大(0.4347),也说明o-M-CH2的Rh—O键共价作用最弱,易于断开,是最有利于决速步骤的催化剂。
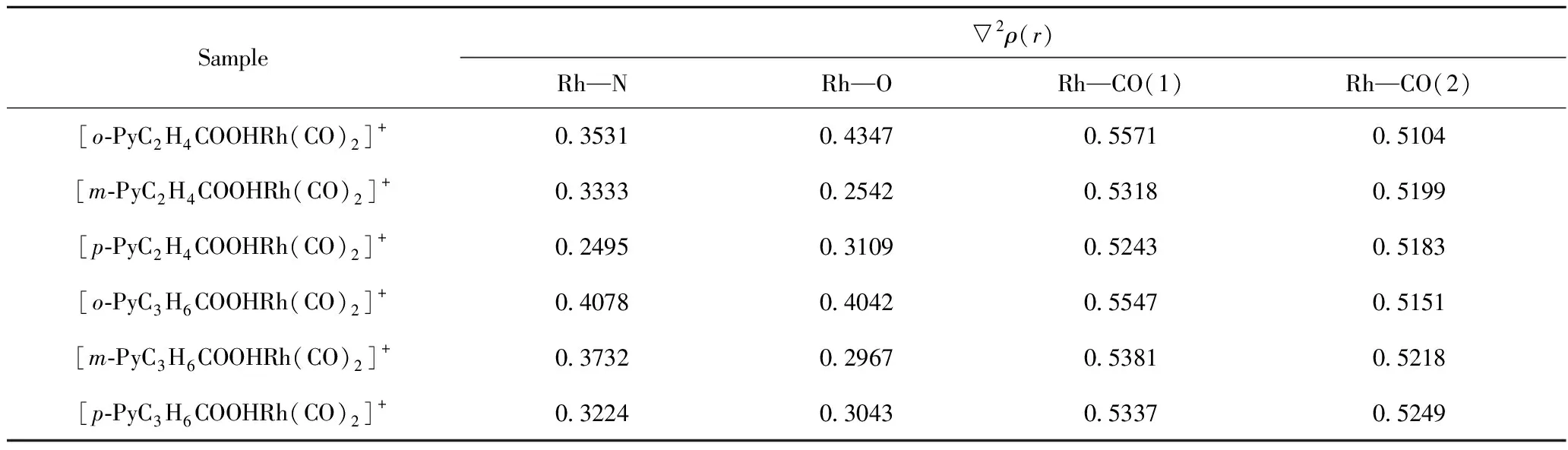
表2 [Rh(CO)2I2)]-、羧基吡啶铑阳离子系列催化剂中心原子Rh与周围原子间键鞍点电子密度的Laplacian量▽2ρ(r)Table 2 Electronic density and Laplacian (▽2ρ(r)) of critical points in Rh catalysts
2.3 催化反应的过渡态和能垒
CH3I氧化加成反应是Rh(I)催化甲醇羰基化反应中的决速步骤,图4为[Rh(CO)2I2)]-和[o-PyC2H4COOHRh(CO)2]+与CH3I氧化加成反应能垒示意图。所有反应过渡态经振动分析证实拥有唯一虚频,能量经零点能校正。结果表明,[Rh(CO)2I2)]-与CH3I氧化加成反应,经过渡态TS1,反应能垒103.6 kJ/mol;包含吡啶、羧酸给电子基团的[o-PyC2H4COOHRh(CO)2]+,CH3I加成反应能垒更低(93.5 kJ/mol);该催化剂的不对称螯合结构有效协同了CH3I氧化加成,具有更高活性。对比[o-PyC2H4COOHRh(CO)2]+的CH3I氧化加成反应过渡态和加成产物构型(见图3),可以发现CH3I加成前Rh—N键长为0.2078 nm,过渡态(TS2)Rh—N键长0.2103 nm,加成后Rh—N键长0.2109 nm,整个CH3I氧化加成反应过程中Rh—N键长基本不变,说明Rh—N之间形成了较强的配位键,这种配位作用也提高了[o-PyC2H4COOHRh(CO)2]+催化剂的稳定性。
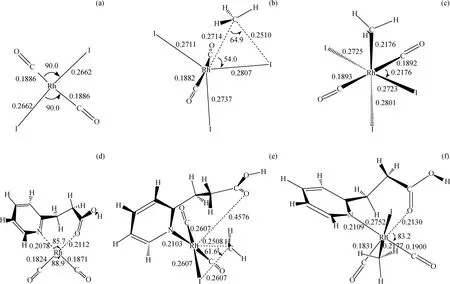
图3 [Rh(CO)2I2)]-和[o-PyC2H4COOHRh(CO)2]+与CH3I氧化加成反应过程中各驻点几何结构Fig.3 Geometric structure of reactants, transition states, intermediates, and products in methanol carbonylation at MP2/SDD level Bond length in nm, and bond angle in degree(a) [Rh(CO)2I2)]-; (b) CH3I addition reaction transition state (TS1) in [Rh(CO)2I2)]-; (c) CH3I addition reaction product ([CH3-Rh(CO)2I3)]-); (d) [o-Py-C2H4COOH-Rh(CO)2]+; (e) CH3I addition reaction transition state (TS2) in [o-Py-C2H4COOH-Rh(CO)2]+; (f) CH3I addition reaction product ([o-PyC2H4COOH-CH3Rh(CO)2I]+)
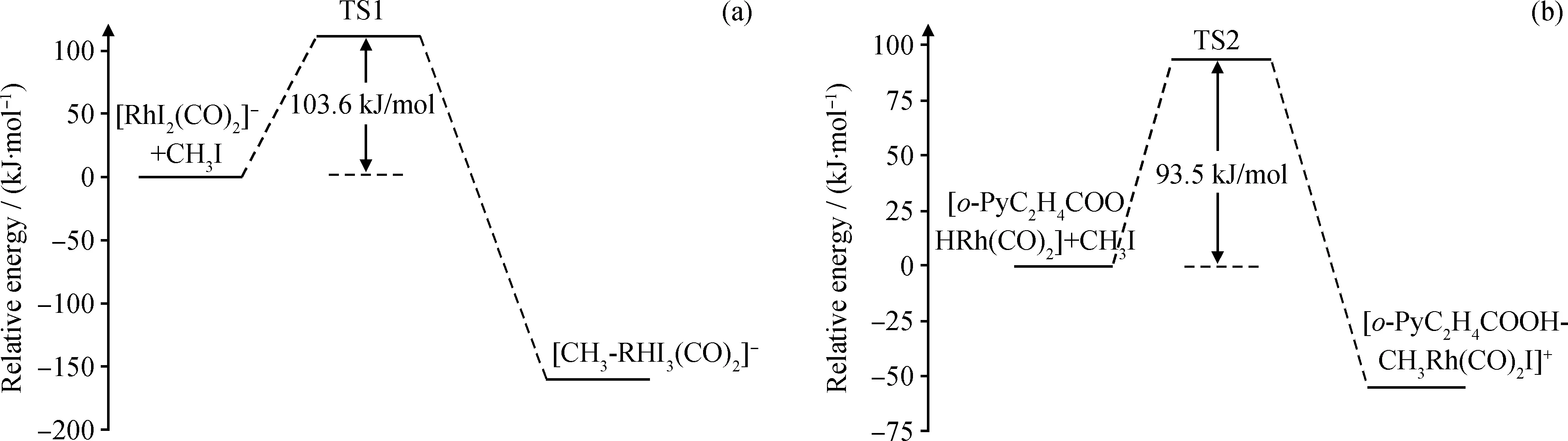
图4 [Rh(CO)2I2)]-和[o-PyC2H4COOHRh(CO)2]+与CH3I氧化加成反应能垒示意图Fig.4 CH3I addition reaction barriers in [Rh(CO)2I2)]- and [o-PyC2H4COOHRh(CO)2]+(a) [Rh(CO)2I2)]-; (b) [o-PyC2H4COOHRh(CO)2]+
3 结 论
(1)包含给电子基团的羧基吡啶铑配合物阳离子催化剂具备更高的反应活性;
(2)Rh—N、Rh—O配位键的不对称双齿配合物催化剂其结构特点是有效协同了CH3I氧化加成,具有更高催化活性,同时Rh—N的配位键增加了催化剂稳定性;
(3)在本文研究的系列配合物催化剂中,邻-吡啶丙酸铑阳离子催化剂具有相对较高的催化活性。
[1] DINGWALL L D, LEE A F, LYNAM J M, et al. Bifunctional organorhodium solid acid catalysts for methanol carbonylation[J].ACS Catalysis, 2012, 2(7): 1368-1376.
[2] WILLIAMS G L, PARKS C M, SMITH C R, et al. Mechanistic study of rhodium/xantphos catalyzed methanol carbonylation[J].Organometallics, 2011, 30(22): 6166-6179.
[3] HOSSEINPOUR V, KAZEMEINI M, MOHAMMADREZAEE A. A study of the water-gas shift reaction in Ru-promoted Ir-catalysed methanol carbonylation utilizing experimental design methodology[J].Chemical Engineering Science, 2011, 66(20): 4798-4806.
[4] ZHANG G, LI Z, ZHENG H, et al. Influence of the surface oxygenated groups of activated carbon on preparation of a nano Cu/AC catalyst and heterogeneous catalysis in the oxidative carbonylation of methanol[J].Applied Catalysis B Environmental, 2015, 179: 95-105.
[5] YAN B, HUANG S, MENGQ, et al. Ordered mesoporous carbons supported Wacker-type catalyst for catalytic oxidative carbonylation[J].AIChE Journal, 2013, 59(10): 3797-3805.
[6] BRODZKI D, LECLERE C, DENISE B, et al. Propriétés catalytiques des complexes des métaux précieux: Carbonylation du méthanol en acide acétique en presence de composes de l’iridium (I)[J].Journal of Molecular Catalysis, 1977, 2(3): 149-161.
[7] VALLARINO L M. Preparation and properties of a series of Halocarbonylrhodates[J].Inorganic Chemistry, 2002, 4(2): 161-165.
[8] HAYNES A, MCNISH J, PEARSON J M.Cis-trans, isomerism in [M(CO)2I4]-, (M=Rh, Ir): Kinetic, mechanistic and spectroscopic studies [J].Journal of Organometallic Chemistry, 1998, 551(1-2): 339-347.
[9] ZHANG S, QIAN Q, YUAN G. Promoting effect of transition metal salts on rhodium catalyzed methanol carbonylation[J].Catalysis Communications, 2006, 7(11): 885-888.
[10] BORAH B J, DEB B, SARMAH P P, et al. Synthesis, reactivities and catalytic carbonylation of rhodium(I) carbonyl complexes containing isomeric acetylpyridine ligands[J].Inorganica Chimica Acta, 2011, 370(1): 117-121.
[11] 雷鸣, 冯文林, 郝茂荣. 甲醇羰基化制乙酸反应的理论研究[J].中国科学(B), 2001, 31(5): 462-467.(LEI Ming, FENG Wenlin, HAO Maorong, Theoretical study on the carbonylation of methanol to acetic acid[J].Sci China Ser, B, 2001, 31(5): 462-467.)
[12] FORSTER D. ChemInform Abstract: On the mechanism of a Rhodium-complex-catalyzed carbonylation of methanol to acetic acid[J].Chemischer Informationsdienst, 1976, 7(17): 846-848.
[13] FLORES-ESCAMILLA G A, FIERRO-GONZALEZ J C. Participation of linear methoxy species bonded to Ti4+sites in the methanol carbonylation catalyzed by TiO2supported rhodium: An infrared investigation[J].Journal of Molecular Catalysis A Chemical, 2012, 359(6): 49-56.
[14] KWAK J H, DAGLE R, TUSTIN G C, et al. Molecular active sites in heterogeneous Ir-La/C-catalyzed carbonylation of methanol to acetates[J].Journal of Physical Chemistry Letters, 2014, 5(3): 566-572.
[15] 吉文欣, 刘翔宇, 冀永强. 吡啶甲酸铑阳离子催化甲醇羰基化反应机理的理论计算[J].催化学报, 2009, 30(11): 1096-1100.(JI Wenxin, LIU Xiangyu, JI Yongqiang. Theoretical calculation for reaction mechanism of methanol carbonylation over pyridine carbonylic acid rhodium cation[J].Chinese Journal of Catalysis, 2009, 30(11): 1096-1100.)
