基于全基因组和准种序列分析3株HAV流行株基因特征
2018-05-24黄腾达周文亭曹经瑗毕胜利
黄腾达 周文亭 曹经瑗 毕胜利
102206 北京,中国疾病预防控制中心病毒病预防控制所
甲型肝炎病毒(hepatitis A virus, HAV)是引起急性甲型肝炎的病原体,通过污染的水和食物及人群日常接触经粪口途径传播。据世界卫生组织估计,全球每年约有150万HAV感染病例[1]。HAV属小RNA病毒科,肝病毒属,基因组全长约7.5 kb,为单股正链RNA,包括5′非翻译区、3′非翻译区和一个开放读码框(opening reading frame, ORF),编码2 227个氨基酸残基的多聚蛋白,经蛋白酶裂解为P1、P2及P3。其中P1区编码结构蛋白VP1-VP4,P2和P3区编码非结构蛋白,进一步水解为2 A、2B、2C和3A、3B、3C、3D。根据VP1-2 A连接区的核苷酸序列差异,将HAV分为六个基因型(I-VI)。感染人的HAV主要为I、II、III型,进一步分为A、B两个亚型[1]。基因分型研究表明,我国流行的HAV为基因I型,其中绝大部分属IA亚型[2]。目前HAV只有一种血清型,结构蛋白区氨基酸序列十分保守,其抗原中和位点主要位于结构蛋白VP3和VP1区[3]。
由于病毒RNA缺乏校正功能,在复制过程中形成同源性很高的基因序列群体,即准种(quasispecies)[4]。HAV准种及全序列分析将有利于甲肝病毒暴发流行的溯源研究。本研究对我国某地3株病毒的VP1-2 A区分型一致的序列进行了分析,成功扩增了HAV病毒近全长序列,并进一步分析准种特征,以了解相关序列的遗传特征。
1 材料与方法
1.1标本来源及试剂收集2014年我国某地抗HAV-IgM阳性甲肝急性期患者血清标本3份(KH6.14、KH7.14、KH8.14),标本采集获患者知情同意。抗HAV-IgM试剂盒购自北京万泰公司;病毒核酸提取、质粒小提试剂盒购自Omega公司;DNA胶回收试剂盒购自QIAgen公司,RNasin购自Promega公司,逆转录酶SuperScript III购自Invitrogen公司;Premix Taq购自TaKaRa公司。
1.2引物设计和合成根据HAV全长序列,分9段设计18对引物。其中5′-UTR区分两段分别采用巢式扩增:第一段:F1(1-21):5′-TTCAAGAGGGGTGTCCGGAGT-3′; NR1(470-450): 5′-AATATCCGCCGCTGTTACCCT-3′; F2(18-38): 5′-GGAATTTCCGGAGTCCCTCTT-3′; NR2(451-429): 5′-CTATCCAAGGCATCTCTTCATAG-3′。第二段:NF1(341-361): 5′-CACCTTGCAGTGTTAACTTGG-3′; R1(786-768): 5′-GGTCAAGGCCACTCCCAAC-3′; NF2(367-388): 5′-ATGAACCTCTTTGATCTTCCAC-3′; R2(707-688): 5′-AATGCCCCTGAGTACCTCAG-3′。引物序列位置参照参考株HM175,其余片段引物序列参照文献[5],由华大基因公司合成引物。
1.3病毒核酸提取、逆转录取140 μl血清标本,按试剂盒说明书提取HAV RNA,分别溶解于40 μl焦碳酸二乙酯(DEPC)处理水中。取11 μl RNA模板,加入1 μl(20 μmol/L)外侧下游引物R7或R15[5],1 μl(10 mmol/L)dNTPs,65 ℃加热5 min,取出立即冰浴5 min,按SuperScript III逆转录试剂盒说明书,依次加入5×Frist-Strand Buffer、0.1 M DTT、RNasin和逆转录酶,配制20 μl反应体系,55 ℃水浴1 h,然后70 ℃加热15 min终止反应。
1.4巢式PCR扩增目的片段取5 μl cDNA产物,加入25 μl Premix Taq,外侧上下游引物(10 μmol/L)各2 μl,16 μl DEPC处理水,构成50 μl反应体系,进行一轮PCR,反应条件为:94 ℃ 5 min;94 ℃ 30 s, 50 ℃ 30 s, 72 ℃ 30 s~90 s, 共35个循环;72 ℃ 10 min。取一轮PCR产物2 μl,使用内侧上下游引物,进行二轮PCR反应。1%琼脂糖凝胶电泳鉴定目的条带,由擎科生物公司进行序列测定。
1.5VP1-2A区克隆测序胶回收纯化HAV VP1-2 A区PCR产物片段(nt 2208-3286),按pMD19-T Vector说明书将纯化产物与载体连接,转化到Trans5α大肠埃希菌感受态细胞中,根据蓝白斑筛选原理,每份标本随机挑取30个白斑,用plasmid mini kit进行质粒小提,由擎科生物公司进行序列测定。
1.6基因序列分析使用Lasergene 7.1及BioEdit软件对各片段进行拼接校对及比对。从GenBank下载世界范围内具有代表性的HAV参考株全基因序列13条,我国VP1-2 A连接区分型序列3条。使用Mega7.0软件,Neighbor-Joining法,Kimura-2-parameter模型,Bootstrap值1 000,构建系统进化树。使用RDP4软件进行重组分析, Simplot软件验证结果。使用HyPhy软件包(http://www.datamonkey.com),SLAC法选择最佳核苷酸替换模型,计算dN/dS值,dN和dS分别代表非同义替换率和同义替换率,其中dN/dS>1、dN/dS<1和dN/dS=1分别表明序列处于正向、负向及中性选择[6]。
1.7统计学方法使用SPSS 19.0进行统计学分析,克隆序列中标本内和标本间基因进化距离的比较采用Wilcoxon秩和检验,P<0.05为差异有统计学意义[7]。
核苷酸变异数为各流行株所有克隆序列中与一致序列不同的核苷酸数目,氨基酸变异数为各流行株所有克隆序列中发生非同义替换的氨基酸数目。香农熵值用来反映准种变异特征,其计算公式为:SN/SNA=- [∑i (pi × ln pi)] / ln N,pi为每种独特序列(核苷酸或氨基酸序列)在病毒群体中出现的频率,N为克隆序列总数,香农熵从0(最小多样性)到1(最大多样性)。
2 结果
2.1VP1-2A连接区基因分型及三株流行株序列间同源性分析VP1-2 A连接区(321 bp)系统进化树表明,3株流行株核苷酸、氨基酸序列同源性均为100%,均属于IA亚型,如图1所示。与GenBank中最近的是广西GgGx4.06株,同源性为99.4%;其次是日本AH2株,同源性为99.1%。
3株流行株间全基因组核苷酸同源性为99.9%~100%,存在2~7个核苷酸差异,位于5′端非编码区及非结构蛋白2C、3 A和3D区;全编码区氨基酸同源性为100%。
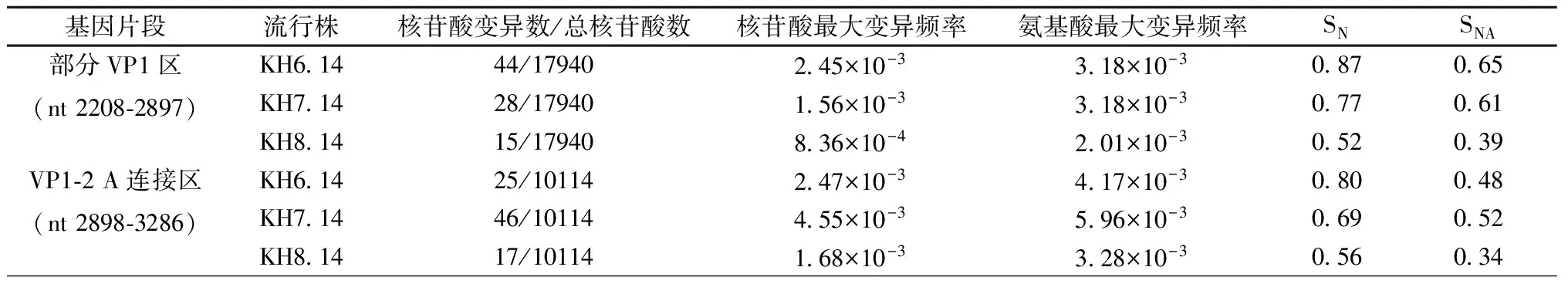
表1 3株流行株在部分VP1区和VP1-2 A连接区准种变异特征Tab.1 Characteristics of quasispecies spectrum in partial VP1 and VP1-2 A region of three strains
2.23株流行株全基因组序列与GenBank中HAV参考株间同源性分析基于全基因组构建系统进化树表明,3株流行株均属于IA亚型,与GenBank中选定的IA亚型参考株全基因组核苷酸同源性为96.0%~98.5%,其中与AH2株亲缘关系最近,同源性为98.5%,全编码区氨基酸同源性为99.0%~99.7%。与选定的其他型别参考株全基因组核苷酸同源性为79.0%~90.7%,全编码区氨基酸同源性为93.1%~98.7%,如图2所示。
2.3抗原中和位点分析文献[8]中报道的抗原中和位点如VP3:Pro65、Asp70、Ser71、Gln74,VP1:Ser102、Asn104、Lys105、Ser114、Val166、Trp170、Val171、Ala176、Lys221、Gln232,以及基于结构预测发现的表位VP2:Thr71和Ala198[9]。本研究中3株流行株序列在已发表的抗原中和位点处未发生氨基酸变异。
2.4重组及选择压力分析分别针对不同基因片段构建系统进化树,流行株与参考株亲缘关系结果并不一致,例如基于5′-UTR区建树,虽然流行株与AH2株在同一分支,但进化距离明显更远,如图3所示;基于2B区建树,流行株与MNA12-130株亲缘关系更近;而基于3C区建树,AH2株与MNA12-130株自成一支(图省略)。但通过RDP4和Simplot分析未发现明显的亚型内重组事件。3株流行株选择压力分析,全编码区与结构蛋白编码区平均dN/dS值分别为0.028和0.015,均未发现正选择位点。
2.5准种特征分析3株流行株序列全长VP1-2 A区克隆测序,共得到78条有效序列(各株为26条序列)。使用所有克隆序列构建系统进化树,序列间呈现无规律的散状分布,如图4所示。每株流行株内的克隆序列,核苷酸和氨基酸同源性分别为99.0%~100%和98.1%~100%;在所有流行株中克隆获得的序列之间,核苷酸和氨基酸同源性分别为99.0%~100%和97.2%~100%。克隆序列中标本内的平均基因进化距离为0.0034,标本间的平均基因进化距离为0.0035,差异无统计学意义(P>0.10)。将序列分为部分VP1区(nt 2208~2897)和VP1-2 A连接区(nt 2898~3286),比较3株流行株各自的核苷酸和氨基酸变异率,VP1-2 A连接区均高于部分VP1区,如表1所示。
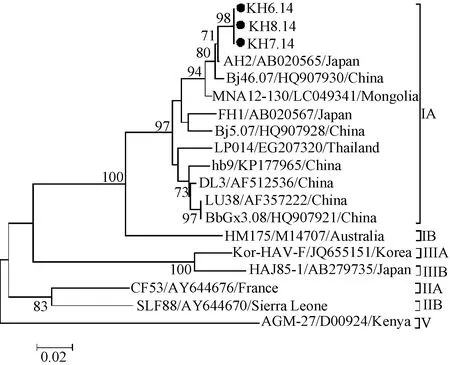
“●”表示本研究中的流行株图1 VP1-2 A连接区核苷酸序列系统进化分析“●”indicates HAV isolates in this studyFig.1 Phylogenetic analysis based on VP1-2 A junction region
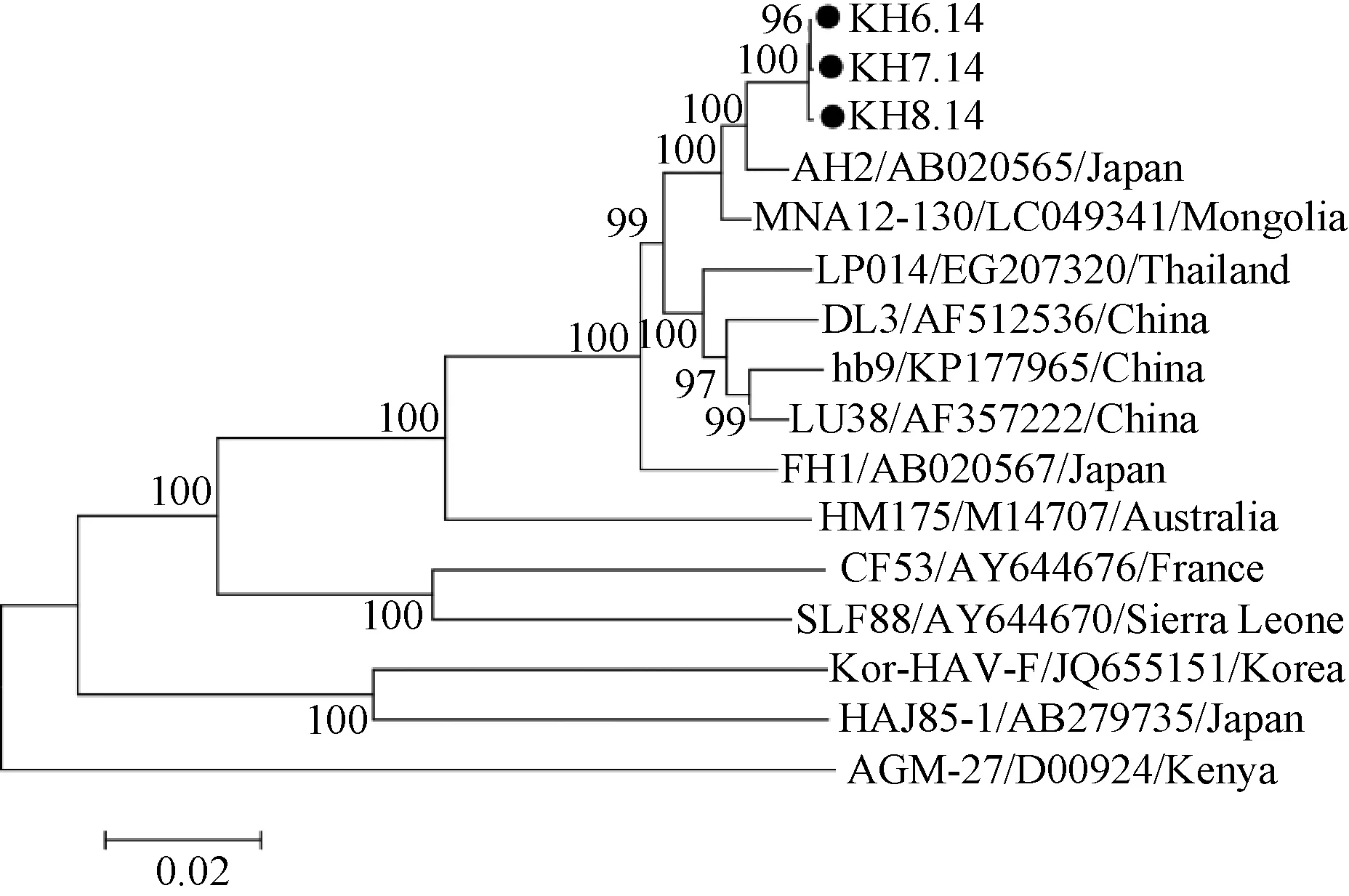
“●”表示本研究中的流行株图2 基于全基因组构建系统进化树“●”indicates HAV isolates in this studyFig.2 Phylogenetic analysis based on whole-genome region

“●”表示本研究中的流行株图3 基于5′-UTR区构建系统进化树“●”indicates HAV isolates in this studyFig.3 Phylogenetic analysis based on 5′-UTR region
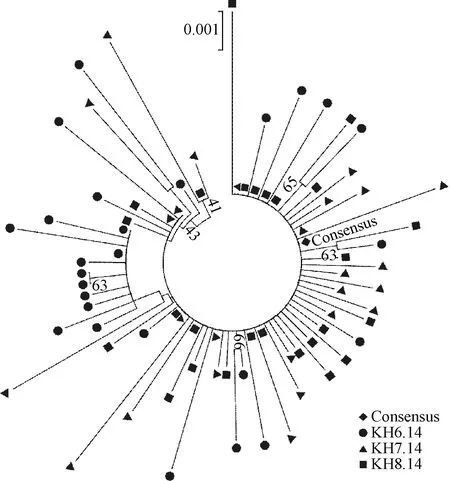
图4 3株流行株克隆VP1-2 A区序列系统进化分析Fig.4 Phylogenetic analysis of cloning sequences based on VP1-2 A region
3 讨论
随着我国城市化进程的加速,卫生环境的改善以及疫苗的广泛使用,全国甲肝的年发病率已由1990年的52.6/10万,降至2012年的1.82/10万[10]。然而,每年小规模的甲肝散发或暴发事件在各省市仍时有发生,给当地公共卫生和疾病防控工作带来挑战。通过HAV分子流行病学研究,将基因分型与现场流行病学调查相结合,利于甲肝病毒的溯源调查[11]。在所有基因片段中,VP1-2 A连接区常用于基因分型[12],已成功对多起HAV食源性或水源性暴发事件进行了基因分型和传播溯源[13-15]。
本研究的3份甲肝患者血清标本,在VP1-2 A连接区核苷酸同源性100%,均属于IA亚型。病毒全长序列的核苷酸和氨基酸同源性分别为99.9%-100%和100%,即核苷酸差异为0.0%~0.1%。有文献报道[16],在VP1-2 A分型片段完全一致的HAV序列中,同源暴发序列其全基因组核苷酸差异可达到0.31%,而非同源暴发相关的序列则更高,可达到0.42%~0.88%。据此推测本实验3株流行株可能来自同一起感染事件。因此获得HAV全基因组序列,结合现场流行病学调查,能够对同源暴发事件进行精确溯源。
同一年份同一地区的流行株可分属于不同分支,如Bj5.07和Bj46.07处于两个不同的分支;不同年份不同地区的流行株可分布于同一分支,如LU38与BbGx3.06具有完全一致的序列,提示我国甲肝流行的复杂性。HAV只有一个血清型,其抗原中和位点主要分布在结构蛋白VP3和VP1编码区,近年发现VP2编码区的第71位、第198位氨基酸也为抗原中和位点[9]。本研究3株流行株序列在已发表的抗原中和位点处均未发生氨基酸变异。监测流行株的抗原中和位点氨基酸变异情况,对甲肝疫苗的使用具有重要意义。本研究流行株中未检测到重组现象,今后随着GenBank中全序列的不断增多,可进一步验证。
全长VP1-2A区克隆测序分析,各流行株的克隆序列间呈无规律的散状分布。流行株内克隆序列、所有克隆序列间核苷酸与氨基酸同源性均较高(分别为99.0%~100%、98.1%~100%和99.0%~100%、97.2%~100%),且克隆序列中标本内和标本间的平均基因进化距离差异无统计学意义,表明流行株间亲缘关系比较近。流行株在VP1-2A连接区的核苷酸和氨基酸变异率均高于部分VP1区,因而VP1-2A连接区被广泛应用于基因分型。本研究观察到KH8.14在3株流行株中核苷酸和氨基酸变异率、香农熵均最低,这可能是由于病毒在宿主体内经受的免疫压力不同,或标本收集时间不同所致。此外,克隆测序方法选择的准种数量有限也可能产生影响。今后应用新一代测序技术等手段,可增加测序广度和深度,通过分析基因组不同区段的准种特性或不同感染来源的流行株准种特点,结合现场流行病学调查,为甲肝溯源和分子流行病学分析提供更为详尽的信息。
利益冲突:无
参考文献
[1] Vaughan G, Goncalves Rossi LM, Forbi JC, et al. Hepatitis A virus: host interactions, molecular epidemiology and evolution [J]. Infect Genet Evol, 2014, 21(1): 227-243. DOI: 10.1016/j.meegid.2013.10.023.
[2] Cao J, Bi S, Meng Q,et al. Genotyping of acute hepatitis a virus isolatesfrom China, 2003-2008 [J]. J Med Virol, 2011, 83(7): 1134-1141. DOI: 10.1002/jmv.22086.
[3] Nainan OV, Xia G, Vaughan G, et al. Diagnosis of hepatitis a virus infection: a molecular approach [J]. Clin Microbiol Rev, 2006, 19(1): 63-79. DOI: 10.1128/CMR.19.1.63-79.2006.
[4] Sánchez G, Bosch A, Gómez-Mariano G, et al. Evidence for quasispecies distributions in the human hepatitis A virus genome [J]. Virology, 2003, 315(1): 34-42. DOI: 10.1016/s0042-6822(03)00483-5.
[5] 王昊. 我国甲肝病毒流行株全基因组序列分析及准种变异研究 [D]. 中国疾病预防控制中心, 2014.
[6] 王昊, 曹经瑗, 毕胜利. 我国甲型肝炎病毒BJ73株全基因组测定与分析 [J]. 中华实验和临床病毒学杂志, 2014, 28(2): 105-107. DOI: 10. 3760/cma. j. issn. 1003-9279. 2014. 02. 010.
[7] 赵琦, 时丽丽, 蒋岩, 等. Miseq高通量测序平台用于一起疑似经性传播HIV的溯源调查 [J]. 中华预防医学杂志, 2014, 48(6): 471-475. DOI: 10. 3760/cma. j. issn. 0253-9624. 2014. 06. 010.
[8] Pérez-Sautu U,Costafreda MI, Caylà J,et al.Hepatitis A virus vaccine escape variants and potential new serotype emergence [J]. Emerg Infect Dis, 2011, 17(4): 734-737. DOI: 10.3201/eid1704.10.1169.
[9] Wang X, Ren J, Gao Q, et al. Hepatitis A virus and the origins of picornaviruses [J]. Nature, 2015, 517(7532): 85-88. DOI: 10.1038/nature13806.
[10] Xu ZY, Wang XY. Live attenuated hepatitis A vaccines developed in China [J]. Hum Vaccin Immunother, 2014, 10(3): 659-666. DOI: 10.4161/hv.27124.
[11] Nainan OV, Armstrong GL, Han XH, et al. Hepatitis a molecular epidemiology in the United States, 1996-1997: Sources of infection and implications of vaccination policy [J]. J Infec Dis, 2005, 191(6): 957-963. DOI: 10.1086/427992.
[12] Yilmaz H, Karakullukcu A, Turan N, et al. Genotypes of hepatitis a virus in Turkey: first report and clinical profile of children infected with sub-genotypes IA and IIIA [J]. BMC Infect Dis, 2017, 17(1): 561. DOI: 10.1186/s12879-017-2667-3.
[13] Collier MG, Khudyakov YE, Selvage D, et al. Outbreak of hepatitis A in the USA associated with frozen pomegranate arils imported from Turkey: an epidemiological case study[J]. Lancet Infect Dis, 2014,14(10):976-981. DOI: 10.1016/S1473-3099(14)70883-7.
[14] Severi E, Verhoef L, Thornton L, et al. Large and prolonged food-borne multistate hepatitis A outbreak in Europe associated with consumption of frozen berries, 2013 to 2014 [J]. Euro Surveill, 2015, 20(29): 21192. PMID: 26227370.
[15] Bruni R, Taffon S, Equestre M, et al. Key role of sequencing to trace hepatitis A viruses circulating in Italy during a large multi-country European foodborne outbreak in 2013 [J]. PLoS One, 2016, 11(2): e0149642. DOI: 10.1371/journal.pone.0149642.
[16] Vaughan G, Xia G, Forbi JC, et al. Genetic relatedness among hepatitis A virus strains associated with food-borne outbreaks [J]. PLoS One, 2013, 8(11): e74546. DOI: 10.1371/journal.pone.0074546.
