6-氟-4-色满酮-2-甲酸合成的工艺研究
2018-05-19王永辉柏一慧施倩影
王永辉 柏一慧 施倩影
(1.滁州城市职业学院 安徽滁州 239000;2.浙江师范大学化学与生命科学院 浙江金华 321004)
苯并吡喃衍生物由一个苯环和一个吡喃环构成,是一类具有广泛生理活性[1-2]和药理活性的六元杂环化合物,在药物合成中起到重要作用,越来越受到人们的重视[3-6]。
苯并吡喃-4-酮骨架(4-色满酮)是化学药物中的一个基本母核[7-9],可作为多种药效基团的载体部分,而6-氟-4-色满酮-2-羧酸具有色满结构单元,是合成β1-受体拮抗剂药物的中间体之一[10-12],因此,6-氟-4-色满酮-2-羧酸衍生物的合成具有重要意义,应用前景广阔。本文从对氟苯酚和碘甲烷出发,先合成对氟苯甲醚,然后在AlCl3催化下,与顺丁烯二酸酐经傅-克酰基化反应,再环化[13],将4-(5-氟-2-羟基)苯基-4-酮-2,3-丁烯酸溶解于碳酸氢钠溶液中,最后得到白色晶体6-氟-4-色满酮-2-甲酸(收率86.9%)。该方法具有操作简单、收率高、反应时间短的优点。
一、实验部分
(一)主要仪器与试剂。仪器:BD-II多功能紫外分析仪(北京启航博达科技有限公司),WRS-1C熔点测定仪(上海仪电科学仪器股份有限公司),RE-501旋转蒸发仪(上海科升仪器有限公司),AUY120电子分析天平(岛津企业管理(中国)有限公司),SXJQ-1电子数显恒速搅拌器(郑州科华仪器设备有限公司),FTIR-1500红外光谱仪(上海精密仪器仪表有限公司),GC1690气相色谱仪(杭州捷岛科学仪器有限公司),LC-100高效液相色谱仪(上海伍丰科学仪器有限公司),SHB-III真空水泵(天津科诺仪器设备有限公司)。
试剂:4-氟苯酚,分析纯,上海凯赛化工有限公司;其它试剂为分析纯。
(二)实验合成路线。在丙酮浓溶液中,对氟苯酚和碘甲烷反应生成对氟苯甲醚,再与顺丁烯二酸酐反应,经酰基化(B-C)和环合(C-D)过程,最终生成6-氟-4-色满酮-2-甲酸。

(三)实验过程。
1.对氟苯甲醚的制备。用无水硫酸镁干燥丙酮过夜备用,取14.949g对氟苯酚溶解在80ml干燥过的丙酮中,再加入27.844gCH3I、73.968gK2CO3加热回流约2h,后过滤减压蒸馏收集馏分6.369g,并测其薄层色谱中(石油醚:乙酸乙酯=3:1)的比移值为0.75。
2.4-(5-氟-2-羟基)苯基-4-酮-2,3-丁烯酸的制备。取无水顺丁烯二酸酐固体5.686g放入100ml干燥的烧瓶(上加回流管和干燥管),加入无水AlCl3固体15.620g,再加入50ml二氯甲烷搅拌、加热回流15min(40oC),使其大部分溶解,溶解后溶液呈现黄色。冷却后从滴液漏斗滴中加4.120g对氟苯甲醚,边滴边搅拌。滴加完加热回流,用PH试纸在干燥管口测试放出气体酸碱性。至不再放出酸性气体,停止加热。此时液体变为深红色(约6h)。
将装置改为蒸馏装置,把二氯甲烷蒸除,冷却后缓慢加入10ml浓盐酸和50ml水混合溶液(冷水浴),放出大量气体,生成黄色固体混有红色油状物。抽滤得到黄色固体6.662g。重结晶、水洗得到黄色固体6.231g,测得其熔点为191-193oC,在乙酸乙酯薄层色谱中比移值为0.65。IR(KBr)ν:1728,1648,1601,1484cm-1;1HNM(DMSO-d6,400MHz,δppm):13.13(1H,-COOH),11.08(1H,-OH),7.91(1H,-CH),7.3-7.54(2H,ArH),7.01(1H,ArH),6.645(1H,-CH)。
3.6-氟-4-色满酮-2-甲酸的制备。取0.727g4-(5-氟-2-羟基)苯基-4-酮-2,3-丁烯酸晶体放入100ml烧瓶,加入50ml6%碳酸氢钠溶液,搅拌下加热回流黄色晶体渐溶,溶液成红棕色后又慢慢变淡,反应14min完毕溶液成无色。冷却后缓慢滴加浓盐酸,调节PH至1,静置,有白色晶体析出,抽滤,水洗,风干后称得晶体重0.677g。在乙醇色谱中的比移值为 0.5,测其熔点为 159-166oC。IR(KBr)ν:3087,3051,2931,1752,1654,1485cm-1;1HNM(CDCl3,400MHz,δppm):12.24(1H,-COOH),7.24-7.55(2H,ArH),7.09-7.12(1H,ArH),5.16(1H,ArOCH),3.05-3.18(2H,ArCOCH2),;MS(EI)m/z:211(M++1),210(M+),165,138。
二、结果与讨论
(一)催化剂浓度对产物4收率的影响。

表1 催化剂浓度对收率的影响
由实验数据结果表明碳酸氢钠溶液的浓度为6%左右反应结果最佳,用量没有特殊要求,因为它是个自身的环合反应,但碳酸氢钠溶液也不能过多只要能将其溶解就可以了。曾用2%NaHCO3溶液加热回流14min,实验结果产物很少。也曾用0.5%NaHCO3溶液加热回流1h没有沉淀生成且在乙醇色谱中有两个点,比移值分别为0.85、0.275,可能是由于碱性不足,反应不完全所致。用4%和12%的碳酸氢钠溶液加热回流至反应完全,虽然也有产物但收率并不理想。实验结果显示6%的碳酸氢钠溶液反应结果最佳,收率高。
(二)反应时间对产物4收率的影响。第三步环合反应需要加热回流,温度控制在100℃以下,反应时间不能过长,以防副反应的发生。实验结果表明反应时间为14min左右反应结果最佳,曾将产物C在碳酸氢钠溶液中回流加热较长时间(2h),仅得微量固体。也曾用5%的NaHCO3溶液在室温下反应七天后加热回流38min,反应结果析出白色晶体很少,且有大量棕褐色杂质生成,可能因为反应时间长产生了副产物。后用6%的NaHCO3溶液用薄层色谱跟踪加热回流14min反应结束,且反应效果较好。所以反应时间不能过长温度也不能太高,保持回流即可。结果如表2所示。
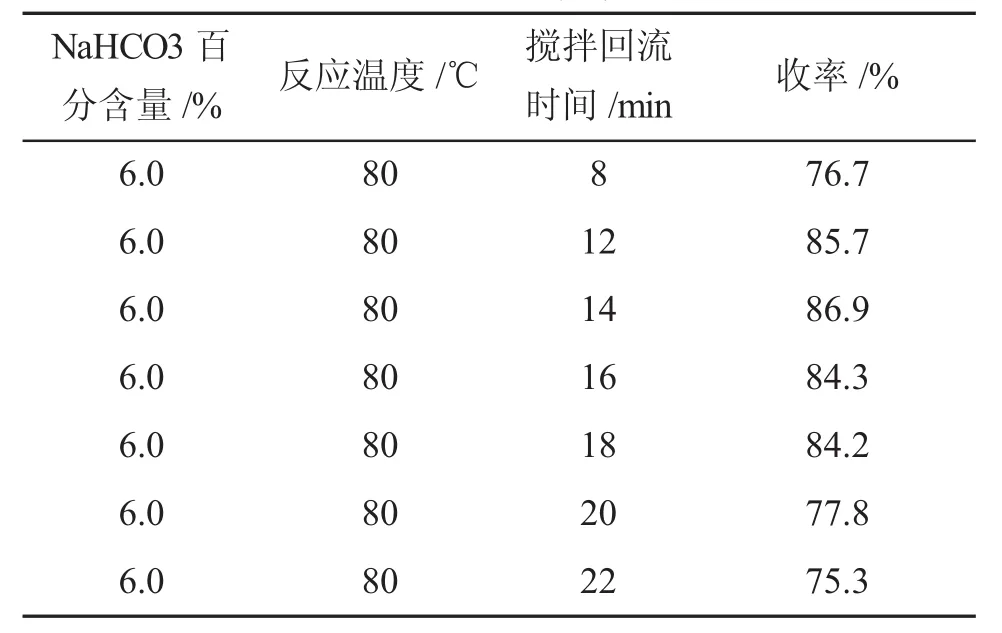
表2 反应时间对收率的影响
反应时间过短,则反应不完全所得产物纯度也不高,如反应时间为8mim产率仅为76.7%,且所得晶体还含有淡黄色;时间太长则导致反应物氧化、收率降低,如表中反应时间大于20min产率较之前的降为77.8%。实验结果表明在回流温度下,回流时间为14min产率最高。
(三)碱的种类对产物4收率的影响。

表3 催化剂种类对收率的影响
在加成环合这一步,催化剂的碱性强弱对反应产生影响,碱性强可使酚氧负离子发生氧化,副产物增多,碱性弱可使酚氧负离子难产生,加成将受阻。用碱性较弱的碳酸氢钠溶液作为反应介质,并通过增加碱的浓度,在回流温度下反应,获得了较好的收率(86.9%),取得了很好的效果。
(四)反应机理。Michael加成反应是亲核加成反应,在相同反应条件下亲核反应的难易主要取决于亲核试剂的亲核能力,而亲核试剂的亲核能力与其电子云密度(碱性)、可极化度以及空间自由度有关。环合反应,在碱性条件下氧原子上的负电荷进攻双键,迫使双键打开,发生1,4-Michael加成,环合得到产物。实验表明,在保证溶解的前提下,碱性条件下环合反应机理如下:
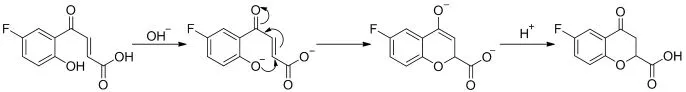
三、结论
通过实验及对实验结果的分析最终得出以下结论:
(一)以4-氟苯酚为原料,经甲基化、傅克酰基化反应、迈克尔加成,合成6-氟-4-色满酮-2-甲酸。
(二)整个合成路线中第三步反应是关键,最佳工艺条件为6%碳酸氢钠溶液作为催化剂,回流温度80℃,反应时间14min,产量为86.9%。
参考文献:
[1]HarborneJB,MabryTJ,MabryH.Theflavonoidsaolvances inresearch[M].London:ChapmanandHall,1982:15.
[2]张惠斌,爱起利,黄文龙.苯并吡喃酯类化合物的合成及其生物活性[J].中国药物化学杂志,2000,10(3):157-160.
[3]XhonneuxRM,VanLommenGE.Agentsforloweringthe bloodpressure[P].EP334429,1989.
[4]王桂霞,张洲洋,刘鹏,等.具有生物活性的4-色满酮衍生物的合成研究进展[J].化学通报,2017(4):322-328.
[5]Kurono M,Kondo Y.Hydantion derivatives for treating complications of Diabetes[P].EP0264586,1989.
[6]李慧媛,胡春.4-色酮类和4-色满酮类化合物生物活性研究概况[J].中国药学杂志,2005,40(4):241-244.
[7]周树宝.6-氟-2-取代-4-色满酮的合成研究[D].新疆:新疆大学,2010.
[8]陆昆,孟岩.关于6-氟4-色满酮-2-羧酸的合成分析[J].赤峰学院学报,2017(21):9-10.
[9]张毅文.6,7-亚甲二氧基-4-色满酮衍生物的合成及生物活性研究[D].陕西:西北农林科技大学,2013.
[10]王树香,李术娜,李红亚,等.3-芳亚甲基色满酮类化合物的研究进展[J].化学试剂,2014(7):609-616.
[11]李洪爽,张瑞泽,郭爽.基于选择性羟基保护合成3-烷基-5,7-二羟基-6-氯-4-色满酮 [J].化学试剂,2017(3):321-325.
[12]马虹,刘志军.β受体阻滞剂在治疗高血压病中的地位[J].中华心血管病杂志,2006,34(7):660-661.
[13]Shiratsuchi M,Kawamura K,Akashi T,et al.Synthesis and Hypotensive Activity ofBenzopyran Derivatives[J].Chem PharmBull,1987,35(2):632-641.
