无油樟与葡萄、拟南芥、水稻基因组的多倍化及共线性分析
2018-05-18王振怡潘玉欣
张 岚,袁 敏,王振怡,潘玉欣
(华北理工大学生命科学学院/华北理工大学基因组学与计算生物学研究中心,河北唐山063210)
全基因组加倍,又称伴随基因丢失的多倍化,长期以来已经成为动物、真菌和其他生物,尤其是植物基因组进化的一个重要驱动力[1]。随着全基因组加倍的进行,基因组结构常常会发生明显改变,如染色体重组、基因倒位、基因丢失等,而多倍化产生的大量重复基因成为遗传创新的重要材料来源[2]。被子植物是迄今为止陆地植物的最大族群,有超过300 000种生物,大多数开花植物的进化过程均受到多倍化影响[3]。研究表明,在被子植物的早期进化过程中甚至存在古多倍化事件。无油樟是纵观被子植物历史的一个重要的参照,是已测序的开花植物中最古老的物种之一,基因组结构和种系基因组学分析表明,被子植物的祖先是多倍体,祖先基因在被子植物生物学中起着关键作用[4-6]。双子叶植物的共同祖先有7条染色体,在13 000万年前经历了双子叶植物共有的三倍乘(γ)事件后,又经历染色体的融合等过程,葡萄基因组形成现有的19条染色体[7-8]。作为首先被测序完成的开花植物,拟南芥在和葡萄一样经历了双子叶植物共同祖先的三倍乘(γ)事件后,又发生了最近的 2次二倍乘(α,β)事件[9-12]。水稻是单子叶禾本科植物中重要的模式植物,基因组拼接较为完整,其结构也最接近禾本科植物基因组。水稻基因组在约1亿年前经历了禾本科植物共有的全基因组加倍事件,而后又先后发生了2次自己独立的全基因组二倍乘事件[13-14]。这些多倍化产生的重复基因为大范围分析基因进化和功能提供了有价值的参考。
本研究对无油樟、葡萄、拟南芥、水稻基因组进行比较基因组学分析,通过4个物种基因组内和基因组间的同源共线性分析,明确多倍化过程对重复基因造成的影响,解析基因组在进化过程中发生的变化,对于探寻单、双子叶植物的共同起源以及研究被子植物的进化过程具有重要的意义。
1 材料与方法
1.1 全基因组序列数据
葡萄(Vitis vinifera,简称 Vv)的全基因组序列来自Phytozome(https://phytozome.jgi.doe.gov),无油樟(Amborella trichopoda,简称 Ar)、拟南芥(Arabidopsis thaliana,简称 At)和水稻(Oryza sativa,简称 Os)的全基因组数据来自 PGDD(http://chibba.agtec.uga.edu/duplication)。其中,无油樟基因组涉及1 210个scaffolds,26 846个基因;葡萄有19条染色体,26 346个基因;拟南芥有5条染色体,27 416个基因;水稻有12条染色体,39 049个基因。
1.2 全基因组同源结构分析
为了更准确地寻找各物种基因组内以及基因组间的同源结构片段,本研究对获取的原始基因组数据进行了数据预处理。对于无油樟,选取前100个长度2 Mb以上的scaffolds,其他物种均去除没有锚定到染色体上的基因。利用基本局部序列比对工具 BLASTP(E≤10-5)[15],对单一物种基因组内以及2个物种基因组间作双序列比对,获取基因组内以及基因组间的同源基因对。
结合基因组内及基因组间双序列比对结果与各个物种染色体的长度信息、染色体上各个基因的位置信息,编写Perl程序绘制同源结构点阵图,并且标记出基因对间匹配最好、次好以及其余的3个基因对。根据同源结构点阵图上基因对组成的成线性的片段,验证各个物种在进化历程中发生的全基因组加倍事件,同时为共线性数据分析提供图示化依据。
1.3 全基因组共线性分析
利用共线性分析工具 MCScan[16]和 Colinearscan(P<0.05)[14],获取基因组内以及基因组间同源共线区域。为了描述同源共线片段的相似性,在基因组内和基因组间,考虑到大的基因家族(重复基因对数≤30个)[17]以及串联重复基因(基因对间相对位置 <2×106bp)[18]的影响,按照同源共线区域内包含的基因对数量,统计在不同长度范围的同源共线区域的数量和区域内包含的同源共线基因对数量、无油樟与其他物种基因组间共线性基因的保留情况,以及无油樟与拟南芥、葡萄、水稻基因组间共有的无油樟基因占共线性基因的百分比。
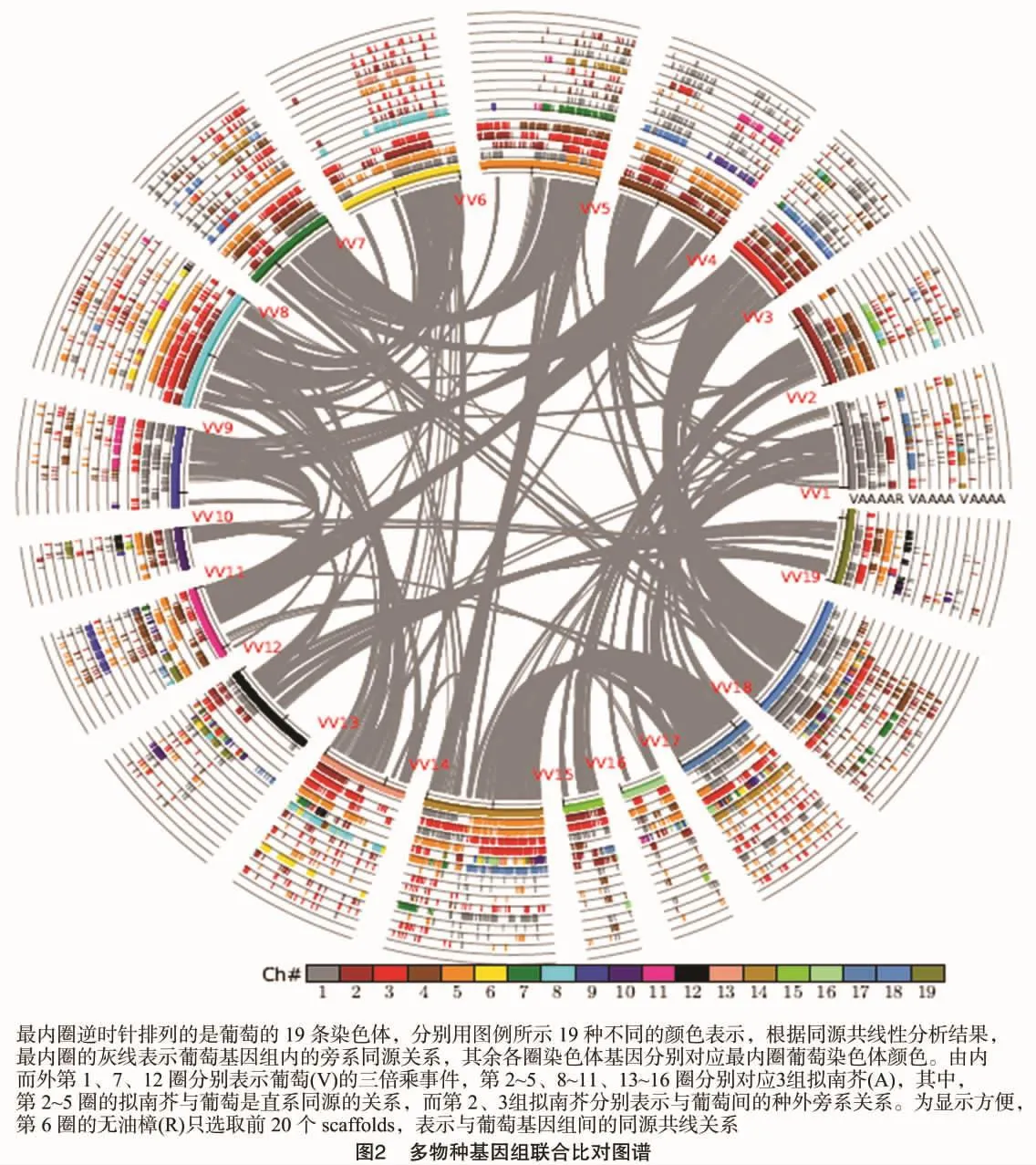
1.4 多物种基因组联合比对图谱的构建
本研究以葡萄基因组三倍乘事件的共线性基因作为参考列,结合基因组内和基因组间同源共线性分析结果,建立多物种基因组联合比对列表,并编写python程序,利用Matplotlib绘制多物种基因组联合比对图谱,以圈图的形式反映基因组间的相似性。
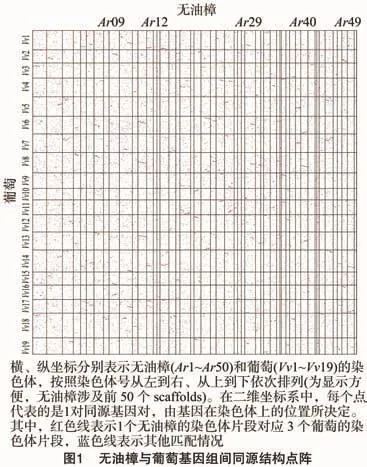
2 结果与分析
2.1 基因组内及基因组间同源结构分析
以1个已知的较为古老的物种或者全基因组发生加倍次数较多的物种基因组内的同源结构点阵图为依据,结合2个物种基因组间的同源结构点阵图,可以基本推断后一物种在进化过程中在全基因组层面发生的主要加倍事件,而且能够统计出在不同加倍事件中参与的基因数量。
在同源结构点阵图中,红色的点表示在物种基因组间进行的蛋白质序列比对中,处于最佳匹配的同源基因对,表示物种间的直系同源片段;蓝色的点表示的是次好匹配的同源基因对,而灰色的点表示的是其余的同源基因对,均表示种间旁系同源片断。
无油樟是被子植物中已测序的最古老的物种,其基因组没有被锚定到某一条染色体上,从无油樟基因组内的同源结构点阵图中,看到的是一些非常短小的线段,很难分析出其自身的加倍过程。而在无油樟和葡萄2个物种基因组间的同源结构分析中,发现无油樟的9号scaffold与葡萄的2、5、16号染色体之间存在1∶3同源共线关系,无油樟的29号scaffold与葡萄的6、8、13号染色体之间存在的1∶3同源共线关系。在无油樟的第12、40号scaffold与葡萄的6、8、13号染色体之间,以及第49号scaffold与葡萄的第1、14、17号染色体间也存在明显的1∶3的同源共线关系。同样,在其他scaffold与葡萄染色体之间也存在类似情况(图1)。这与 Amborella Genome Project报道的结果[4]相符。
2.2 同源共线片段推断
本研究使用共线性分析工具推测无油樟与葡萄、拟南芥、水稻基因组内和基因组间的同源共线片段。结果表明,同源性往往依赖于长的同源共线性片段,而不同基因组间的高相似性对解析基因组结构、功能与基因家族的分析提供了帮助[18]。
在无油樟基因组内,存在102个长度≥4个基因对的同源共线区域,包含473个基因对,这说明其同源共线性较差。同样,在同源结构点阵图中也看不到大段的连续区域。当同源共线区域≥20个基因对长度时,葡萄基因组内部包含21个同源共线区域,595个同源共线基因对,最长的片段位于葡萄的5号和7号染色体之间,包含52个基因对;拟南芥基因组内部包含52个同源共线区域,有2 472个同源共线基因对,最长的片段位于拟南芥的3号和2号染色体之间,包含181个基因对;水稻基因组内包含34个同源共线区域,共2 774个同源共线基因对,最长的片段位于水稻的1号和5号染色体之间,包含250个基因对。而在水稻基因组内,在不同的基因对长度范围,得到的同源共线性区域以及同源共线性基因对的数量相对较多,超过200个基因对长度的区域有2个,包含499个同源基因对。大量的长片段存在于水稻和拟南芥基因组内(表1),这与同源结构点阵图得到的结论相符。研究表明,无油樟与其他几个物种相比,拥有最短的共线性同源区域,它是最古老的物种,在漫长的历史进程中,基因组的结构遭受了严重的破坏,而其他几种模式植物则相对保存了较好的同源共线性。
在无油樟基因组与其他物种的共线性分析中,发现在不同长度同源共线区域限定时,无油樟与葡萄的基因组间保存相对较好的同源共线区域及同源共线基因对数量。当同源共线区域≥10个基因对长度时,在无油樟与葡萄基因组间有139个同源共线区块,包含2 311个基因对;无油樟与拟南芥基因组间包含14个同源共线区块,包含164个基因对;在无油樟与水稻基因组间包含49个同源共线区块,包含622个基因对。当同源共线区域≥20个基因对长度时,在无油樟与葡萄基因组间有33个共线性区块,包含928个基因对,最长的片段位于无油樟的第2号scaffold和葡萄的第5号染色体之间,包含52个基因对;在无油樟和水稻基因组间包含2个共线性区块,包含42个基因对,最长的片段位于无油樟的第25号scaffold和水稻的第2号染色体之间,包含22个基因对;无油樟和拟南芥基因组间没有发现超过20个同源共线基因对的同源共线区域。在无油樟与葡萄间甚至存在2个超过50个基因对长度的区域,共有102个同源共线基因对(表1)。研究发现,以只经历了古老的被子植物全基因组加倍事件的无油樟基因组为参考,水稻和拟南芥基因组在进化历程中经历的加倍事件相对葡萄基因组复杂,二者所保留的同源共线区域和同源共线基因对数比葡萄基因组少。
2.3 同源共线性基因的保留情况分析
本研究涉及的4个物种在经历了各自的加倍事件后,其基因组不可避免地发生了分离、融合现象,从而导致各物种的基因存在不同程度的丢失,其中葡萄基因组丢失得最少(49.1%),水稻的次之(62.1%),拟南芥丢失得最为严重(652%)。这可能是由于葡萄有19条染色体,其基因组保留了大量的祖先基因,基因组比较稳定。在葡萄、拟南芥、水稻3个物种之间,拟南芥和葡萄间保留的同源共线性基因比例达到了40.8%,而水稻和葡萄、水稻和拟南芥之间保留的同源共线性基因则相对较少,显然这与被子植物单、双子叶间的亲缘关系相关,亲缘关系越近,同源共线性基因丢失得越少(表2)。
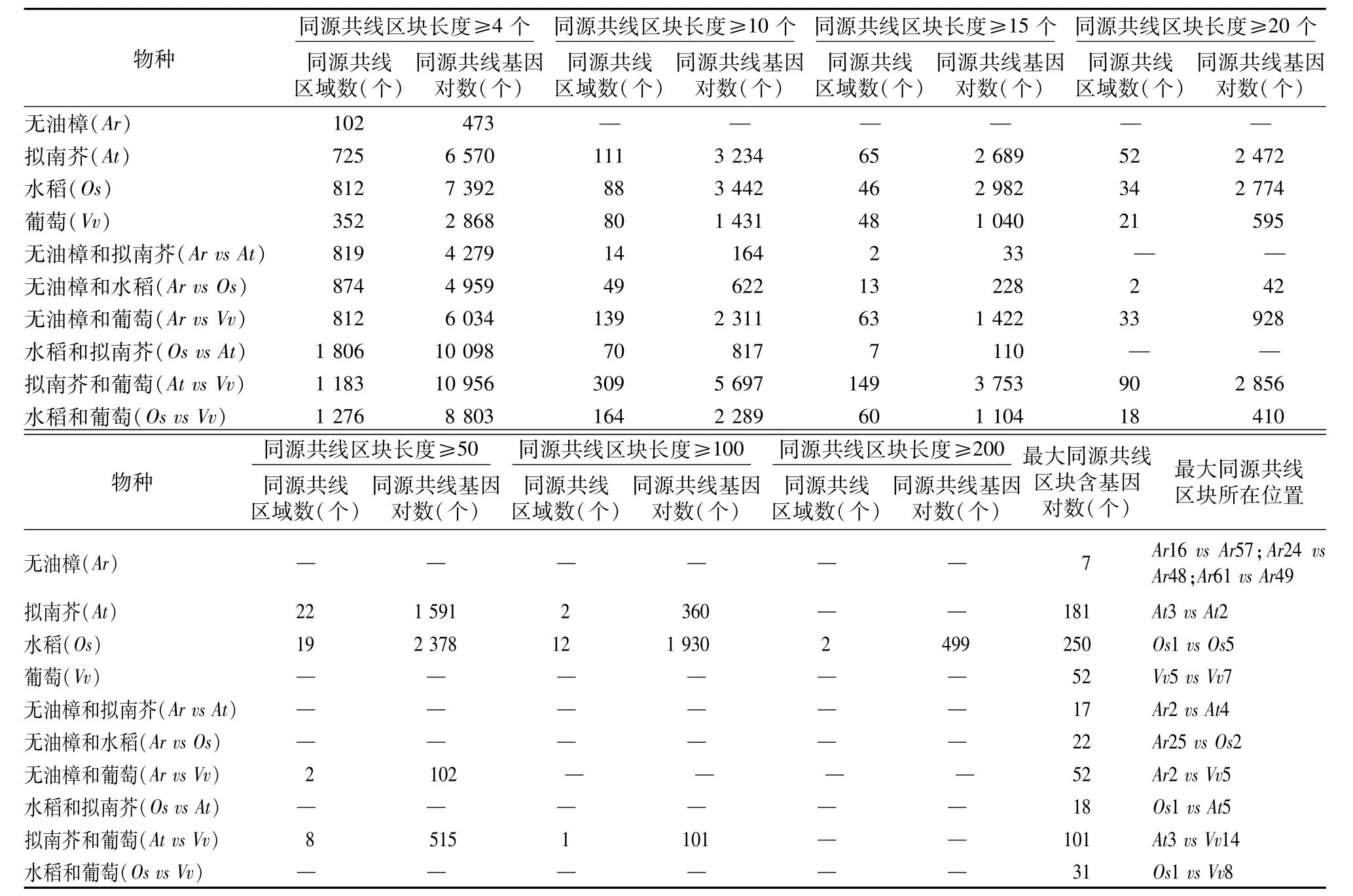
表1 无油樟(Sca1~Sca100)与其他物种基因组内与基因组间同源共线性区域以及基因对数量统计结果
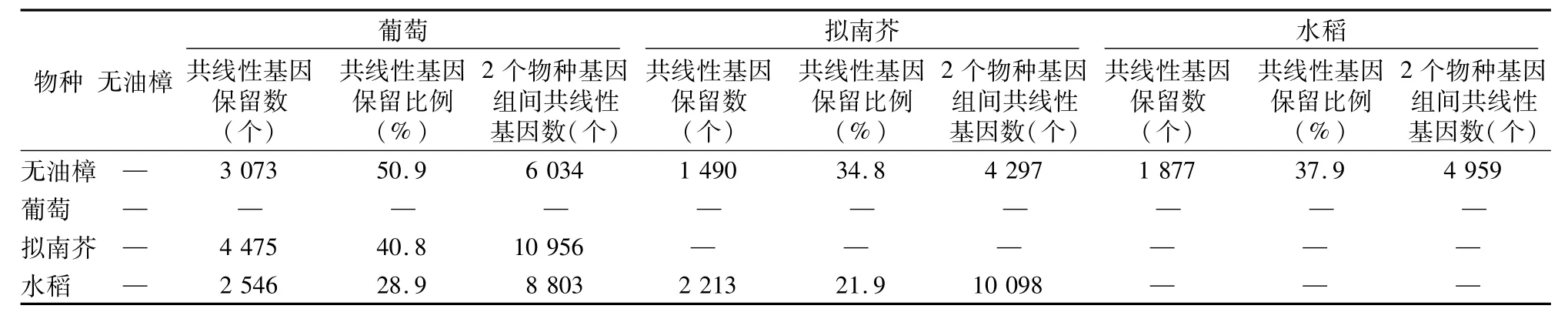
表2 无油樟、葡萄、拟南芥和水稻基因组间同源共线性基因保留情况统计结果
本研究统计了无油樟和拟南芥与无油樟和水稻、无油樟和拟南芥与无油樟和葡萄、无油樟和葡萄与无油樟和水稻保留的无油樟的基因数。发现无油樟和葡萄与无油樟和水稻间保留的无油樟基因数最多(30.6%),无油樟和拟南芥与无油樟和水稻间保留的无油樟的基因数最少(26.7%),详见表3,这说明被子植物的祖先基因在进化过程中发生了大规模丢失,可能是由于物种各自的加倍事件造成的。
2.4 多物种基因组联合比对图谱的构建
作为双子叶植物中重要的模式植物,葡萄基因组保存了双子叶植物最完整的γ事件,在13 000万年前经历了双子叶植物共同祖先的全基因组三倍乘(γ)事件,而后并未发生单独的多倍化事件,因此以葡萄为参考,基于物种基因组内和基因组间的同源共线性分析结果,构建多物种基因组联合比对图谱。
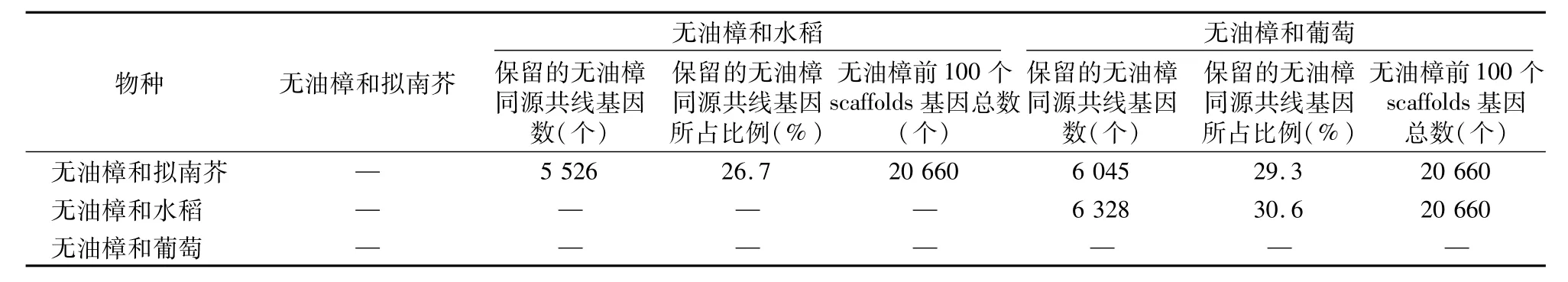
表3 无油樟与葡萄、拟南芥、水稻基因组间保留的Amborella同源共线基因保留情况统计结果
由于葡萄和拟南芥共同经历了双子叶植物共同祖先的三倍乘事件,而拟南芥又先后发生了自己的2次二倍乘事件,因此,图2中的3圈葡萄分别对应3组拟南芥,而每组拟南芥又分别对应自己的2次二倍乘事件。由图2还可以看出,由内往外圈颜色分布越来越淡,这说明物种间的直系同源关系在进化过程中保持了相对完整,而物种内的旁系同源基因对则发生了大规模的丢失。由于无油樟与葡萄间存在1∶3的共线性关系,因此对应有1圈无油樟,可以看出图2在此圈有大量空白,说明存在大量基因片段的缺失。在多物种基因组联合比对图谱中,可以根据不同颜色来判定同源基因在物种间的分布情况,还可以用于分析各物种染色体的重组现象,其中每条染色体上不同的颜色区域都预示可能的染色体重组现象。图谱直观地阐释了各个物种基因组在进化过程中发生的基因丢失情况,同时也反映各物种染色体的结构变化。物种基因组的加倍规模和次数对基因组的影响在图谱中清晰可见,重复基因越多,可能造成基因组的不稳定以及基因丢失的增多。
3 讨论与结论
多倍化是被子植物进化史上的重要特征,全基因组加倍对基因组结构及基因家族的进化有重要的影响[19]。本研究对无油樟、葡萄、拟南芥和水稻基因组进行了比较基因组学分析,在同源基因点阵图中发现无油樟基因组内的同源性较差,而在无油樟与其他几个物种的点阵图中也得到类似结果。而由无油樟与葡萄的点阵图得到与已有报道相同的结果,结构分析表明,无油樟和葡萄基因组之间存在很明显的1∶3的关系,这与古多倍体的γ事件相契合,表明无油樟发生的全基因组加倍不是特殊的谱系事件,可能是发生在2个物种的共同祖先中的,由此推断无油樟的分化要早于古γ事件。这可能是受无油樟基因组测序质量的影响,并与无油樟是已测序的最古老的开花植物有关[4]。
本研究在推测同源共线片段时,采用了严格的阈值限制,包括合适的E值、P值、删除可能的大的重复基因家族和串联重复基因,同时区分同源共线片段的大小,从而保证了研究结果的可靠性[17-18,20]。本研究结果表明,在与无油樟的比较分析中,拟南芥和水稻同源共线性区域以及基因对数都比葡萄少,葡萄基因组所拥有的大片段数也是最多的,这说明物种基因组的加倍次数越多,加倍情况越复杂,所拥有的同源共线区域以及基因对数就越少,拥有的祖先基因也就越少。同时,在2个物种基因组间双向BLASTp得到的最佳匹配基因在共线基因的保留情况统计结果中[19],发现葡萄基因组所保留的基因数最多(50.9%),无油樟与拟南芥、水稻间丢失的无油樟的基因数最多(73.3%),无油樟与葡萄、水稻间丢失的无油樟的基因数最少(69.4%),整体来说,保留的无油樟基因数在1/3左右。本研究结果表明,在经历了物种基因组各自共有和单独加倍事件后,基因组的稳定性明显降低,并伴随着基因的丢失,而丢失仍将继续,该过程大多存在随机性。实际上,从多倍化的推断、非正常重组的分析、基因家族的进化分析等,在很大程度上都依赖于共线性分析,可以说,物种基因组内和基因组间共线性分析是比较基因组研究最重要的内容之一,是解析基因组结构和功能、研究基因、基因家族,以及调控路径等进化规律的重要契机,对基因组学许多研究工作都发挥着至关重要的作用。
参考文献:
[1]Edger P P,Pires JC.Gene and genome duplications:the impact of dosagesensitivity on the fate of nuclear genes[J].Chromosome Research,2009,17(5):699-717.
[2]Otto S P.The evolutionary consequences of polyploidy[J].Cell,2007,131(3):452-462.
[3]Lyons E,Pedersen B,Kane J,et al.Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya,poplar,and grape:CoGewith rosids[J].Plant Physiology,2008,148(4):1772-1781.
[4]Amborella Genome Project.The Amborella genome and the evolution of flowering plants[J].Science,2013,342(6165):1241089.
[5]Chamala S,Chanderbali A S,Der JP,et al.Assembly and validation of the genome of the nonmodel basal angiosperm Amborella[J].Science,2013,342(6165):1516-1517.
[6]Rice DW,Alverson A J,Richardson A O,etal.Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella[J].Science,2013,342(6165):1468-1473.
[7]Tang H B,Bowers JE,Wang X Y,et al.Synteny and collinearity in plant genomes[J].Science,2008,320(5875):486-488.
[8]Jaillon O,Aury JM,Noel B,et al.The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla[J].Nature,2007,449(7161):463-467.
[9]Vision T J,Brown D G,Tanksley S D.The origins of genomic duplications in Arabidopsis[J].Science,2000,290(5499):2114-2117.
[10]Barker M S,Vogel H,Schranz M E.Paleopolyploidy in the Brassicales:analyses of the cleome transcriptome elucidate the history of genome duplications in Arabidopsis and other Brassicales[J].Genome Biology and Evolution,2009,1(1):391-399.
[11]Jiao Y N,Leebens-Mack J,Ayyampalayam S,et al.A genome triplication associated with early diversification of the core eudicots[J].Genome Biol,2012,13(1):R3.
[12]Kaul S,Koo H L,Jenkins J,Analysis of the genome sequence of the flowering plant Arabidopsis thaliana[J].Nature,2000,408(6814):796-815.
[13]Wang X Y,ShiX L,Hao B L,etal.Duplication and DNA segmental loss in the rice genome:implications for diploidization[J].New Phytologist,2005,165(3):937-946.
[14]Wang X Y,Shi X L,Li Z,et al.Statistical inference of chromosomal homology based on gene colinearity and applications to Arabidopsis and rice[J].BMC Bioinformatics,2006,7(1):447-459.
[15]Altschul S F,Gish W,Miller W,et al.Basic local alignment search tool[J].Journal of Molecular Biology,1990,215(3):403-410.
[16]Tang H B,Wang X Y,Bowers J E,et al.Unraveling ancient hexaploidy through multiply-aligned angioserm gene maps[J].Genome Research,2008,18(12):1944-1954.
[17]Wang X Y,Wang JP,Jin D C,et al.Genome alignment spanning major Poaceae lineages reveals heterogeneous evolutionary rates and alters inferred dates for key evolutionary events[J].Molecular Plant,2015,8(6):885-898.
[18]Wang JP,Yu JX,Sun PC,et al.Comparative genomics analysis of rice and pineapple contributes tounderstand the chromosome number reduction and genomic changes in grasses[J].Frontiers in Genetics,2016,7:174.
[19]Jiao Y N,Wickett N J,Ayyampalayam S,et al.Ancestral polyploidy in seed plants and angiosperms[J].Nature,2011,473(7345):97-100.
[20]Wang X Y,Tang H B,Paterson A H.Seventy million years of concerted evolution of a homoeologous chromosome pair,in parallel,in major Poaceae lineages[J].Plant Cell,2011,23(1):27-37.
