异烟肼致肝细胞损伤过程中组蛋白乙酰化对内质网应激的调控作用
2018-05-15李金凤杜莹李标张一杨李莹淑韩铁生任琦冯福民
李金凤,杜莹,李标,张一杨,李莹淑,韩铁生,任琦,冯福民
(华北理工大学公共卫生学院,河北唐山063000)
异烟肼(INH)是WHO推荐的一线抗结核药物,在抗结核治疗中具有不可替代的作用[1,2],但长期服用可导致药物性肝损伤。流行病学调查表明,长期服用INH的结核患者转氨酶升高发生率为10%~20%[3],其中1%~3%患者可出现严重肝损伤,已成为抗结核治疗终止的重要原因之一[4]。肝细胞损伤后,打破了细胞内组蛋白乙酰化酶和组蛋白去乙酰化酶的动态平衡,干扰了染色质重塑,造成炎性因子(如IL-6)、氧化应激指标(如活性氧)等基因异常表达[5,6]。当受损的肝细胞持续受到INH刺激时,大量未折叠蛋白在内质网腔内堆积,产生内质网应激(ERS)[7]。有研究表明,组蛋白乙酰化酶P300可特异性结合在葡萄糖调节蛋白78(GRP78)基因启动子区,调控其下游ERS相关基因表达,继而参与细胞周期调控[8];非酒精性脂肪肝中组蛋白去乙酰化酶HDAC1水平变化可影响CCAAT/增强子结合蛋白同源蛋白(CHOP)的表达,引起慢性ERS,扰乱肝细胞脂质平衡,诱导肝细胞脂肪变性[9,10]。目前在INH致肝细胞损伤过程中组蛋白乙酰化与ERS是否有关尚不清楚。C646为P300的抑制剂,可通过降低P300表达参与细胞的各种生命活动[11];MS-275可通过抑制HDAC1降低组蛋白乙酰化水平,与基因转录活性有关[12]。因此推测,组蛋白乙酰化可能通过调控ERS参与INH致肝细胞损伤。2017年1~5月,我们在INH致肝细胞损伤模型中分别给予C646或MS-275干预,观察了组蛋白乙酰化对ERS的调控作用,以期为INH致肝细胞损伤的预防提供理论依据。
1 材料与方法
1.1 材料 人正常肝细胞株HL-7702(以下称HL-7702细胞),购自中国科学院上海分院。INH,梯希爱(上海)化成工业发展有限公司;C646,美国Cayman Chemical公司;MS-275,瑞士J&K Scientific Ltd公司。台式高速冷冻离心机,德国Heraeus集团;PCR基因扩增仪,美国Bio-Rad公司;酶标仪,美国Molecular Devices公司;荧光定量RCR仪,美国Applied Biosystems公司。二甲基亚砜,日本东京化成工业株式会社;RPMI 1640,美国Corning cellgro公司;ALT、AST检测试剂盒,南京建成生物工程研究所;TaKaRa反转录试剂盒、SYBR®Premix Ex TaqTMⅡ试剂盒,宝生物工程(大连)有限公司;TRIzol,北京百泰克生物技术有限公司;ELISA试剂盒,中国北京冬歌伟业科技有限公司。
1.2 细胞培养 将HL-7702细胞接种于含10% FBS的RPMI 1640培养液中,制成细胞悬液。取细胞悬液5~7 mL分装于50 mL细胞培养瓶中,置于37 ℃、5% CO2的细胞培养箱中,隔天换液1次,待培养瓶底铺满单层细胞时,按1∶2比例进行传代。取传5代对数生长期细胞进行后续实验。
1.3 肝细胞损伤模型制备及鉴定 参照本课题组前期研究方法制备肝细胞损伤模型[13]。取传5代对数生长期细胞,接种于含RPMI 1640培养液的6孔板,每孔2×105个,37 ℃、5% CO2的细胞培养箱中常规培养。培养24 h,随机将细胞分为观察组、对照组,观察组更换为含INH 1 000 μg/mL的RPMI 1640培养液,对照组更换为新的RPMI 1640培养液,继续培养3 h,收集细胞培养液,1 000×g离心5 min,留取上清液。采用ELISA法检测培养液上清ALT、AST活性。根据ALT、AST检测试剂盒说明绘制标准曲线,并按标准曲线计算ALT、AST活性。结果显示,观察组培养液上清ALT活性为(4.31±0.40)IU/L,AST活性为(5.87±0.54)IU/L,对照组分别为(1.44±0.32)、(4.73±0.59)IU/L。观察组培养液上清ALT、AST活性均明显高于对照组(P均<0.05)。表明观察组已经出现肝细胞损伤。
1.4 细胞分组处理 取传5代对数生长期细胞,接种于含2 mL RPMI 1640培养液的6孔板,每孔2×105个,37 ℃、5% CO2的细胞培养箱中常规培养。培养24 h,随机将细胞分为空白对照组、INH组、C646组、INH+C646组、MS-275组和INH+MS-275组,每组设6个复孔。空白对照组更换为2 mL新的RPMI 1640培养液;INH组更换为2 mL含INH 1 000 μg/mL的RPMI 1640培养液;C646组更换为2 mL含C646 5 μmol/L的RPMI 1640培养液;INH+C646组更换为2 mL含C646 5 μmol/L和INH 1 000 μg/mL的RPMI 1640培养液;MS-275组更换为2 mL含MS-275 5 μmol/L的RPMI 1640培养液;INH+MS-275组更换为含MS-275 5 μmol/L和INH 1 000 μg/mL的RPMI 1640培养液。各组继续培养3 h。
1.5 相关指标观察
1.5.1 肝细胞P300、HDAC1、GRP78、CHOP mRNA表达 采用Real-time PCR法。收集各组处理3 h细胞,采用TRIzol法提取细胞总RNA,酶标仪检测260、280 nm波长处的吸光度(A)值,A260/A280为1.8~2.0,表明提取的总RNA纯度合格。取总RNA 2 μL,按照TaKaRa反转录试剂盒说明将其反转录为cDNA,反应条件:37 ℃ 15 min、85 ℃ 5 s、4 ℃ 1 min。以cDNA为模板进行PCR扩增。引物序列由上海生工生物工程股份有限公司设计合成。P300上游引物:5′-AGAAGATGAAGCGGGTTGTG-3′,下游引物:5′-CAGTTGGTGTCGTTGGAGTG-3′;HDAC1上游引物:5′-GGCATCTGGCTTCTGTTACG-3′,下游引物:5′-CTCGTCATCAATCCCGTCTC-3′;GRP78上游引物:5′-GCACAGACGGGTCATTCCAC-3′,下游引物:5′-TCCTATGTCGCCTTCACTCC-3′;CHOP上游引物:5′-CAGAACCAGCAGAGGTCACA-3′,下游引物:5′-ACCATTCGGTCAATCAGAGC-3′;GAPDH上游引物:5′-GAAGGTCGGAGTCAACGGATT-3′,下游引物:5′-CCTGGAAGATGGTGATGGGAT-3′。PCR反应体系共20 μL:ROX Reference Dye 0.4 μL,SYBR Premix Ex Taq 10 μL,DEPC水6.8 μL,上下游引物各0.4 μL,cDNA 2 μL;反应条件:95 ℃ 30 s,95 ℃ 5 s、60 ℃ 30 s、72 ℃ 30 s共40个循环。以GAPDH为内参,采用2-ΔΔCt法计算目的基因相对表达量。实验重复6次,取平均值。
1.5.2 肝细胞P300、HDAC1、GRP78、CHOP蛋白含量 采用ELISA法。收集各组处理3 h细胞培养液,1 000×g离心5 min,留取培养液上清。根据ELISA试剂盒说明绘制标准曲线,根据标准曲线计算各组P300、HDAC1、GRP78、CHOP蛋白含量。
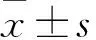
2 结果
2.1 各组P300、HDAC1、GRP78、CHOP mRNA表达比较 见表1。
2.2 各组培养液上清P300、HDAC1、GRP78、CHOP蛋白含量比较 见表2。
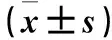
表1 各组P300、HDAC1、GRP78、CHOP mRNA相对表达量比较
注:与空白对照组比较,*P<0.05;与INH组比较,#P<0.05。
表2 各组培养液上清P300、HDAC1、GRP78、CHOP蛋白含量比较
注:与空白对照组比较,*P<0.05;与INH组比较,#P<0.05。
3 讨论
结核病是由结核杆菌引起的、严重威胁人类健康的慢性疾病。据WHO报道,近年来结核病“死灰复燃”,与艾滋病、疟疾一起成为三大传染病。我国是结核病高负担国家之一,WHO推荐的一线抗结核药物INH、利福平、吡嗪酰胺等均对肝脏有不同程度毒性作用,联合用药时肝损害更严重,严重时可发生慢性肝炎、肝纤维化甚至肝衰竭等,常常迫使抗结核治疗中断,继而患者病情加重,同时亦使结核杆菌耐药风险增加。抗结核药物性肝损伤由抗结核药物代谢终产物在体内蓄积引起,破坏了细胞内稳态,导致肝细胞凋亡,继而产生肝损伤,其严重程度因个体差异而不同,目前尚难预测。因此,探讨抗结核药物性肝损伤中的发生机制具有重要意义。
INH在体内主要经肝脏代谢,在肝细胞中经过乙酰转移酶2和细胞色素P450酶系催化,大部分代谢产物经肾脏排出体外,但小部分代谢产物可在体内残留、蓄积,并可直接与肝细胞内大分子结构发生共价结合,改变细胞内组蛋白乙酰化水平,进而诱发肝细胞损伤。组蛋白乙酰化是由组蛋白乙酰化酶P300和组蛋白去乙酰化酶HDAC1共同调控,可通过调控IL-2等转录因子活性,增强细胞内NF-κB等炎性因子表达,继而参与酒精性脂肪肝、肝纤维化等肝脏疾病的发生、发展[14]。P300可结合到组蛋白N端富含赖氨酸残基的尾部,促进染色质转录激活,启动组蛋白乙酰化作用,而HDAC1可使染色质结构致密,抑制基因转录,使组蛋白去乙酰化,二者可相互拮抗。有研究报道,INH能通过影响P300和HDAC1的动态平衡来改变细胞中组蛋白乙酰化状态,从而导致大鼠肝细胞损伤[15]。本研究通过INH处理HL-7702细胞制备肝细胞损伤模型,结果显示,观察组培养液上清ALT、AST含量明显高于对照组,证实INH处理可致肝细胞损伤。本研究结果显示,与空白对照组比较,INH组P300 mRNA表达和蛋白含量均明显降低,HDAC1 mRNA表达和蛋白含量均明显增加,证实组蛋白乙酰化参与INH致肝细胞损伤过程,与本课题组前期研究[15]结果一致。
内质网是真核生物细胞中与分泌相关的重要细胞器,可参与蛋白的合成、转运、折叠和修饰。当外源性或内源性物质刺激细胞时,大量未折叠和错误折叠的蛋白在内质网堆积,影响细胞内钙稳态,诱发ERS。GRP78表达增加会促进细胞存活,而促凋亡分子CHOP表达增加则会促进细胞凋亡,二者作为ERS的标志性分子,在ERS过程中具有重要作用。既往研究发现,ERS与非酒精性脂肪肝、肝硬化等肝脏疾病的发生、发展密切相关[16~19]。槲皮素可升高CHOP mRNA和蛋白表达,激活ERS信号通路,诱导肝星状细胞凋亡[20];糖尿病大鼠经链唑霉素处理后,肝组织GRP78蛋白表达明显升高,未折叠蛋白反应作为ERS的保护性反应,其信号通路被激活,导致肝细胞损伤[21]。本研究结果显示,与空白对照组比较,INH组GRP78、CHOP mRNA表达和蛋白含量均明显升高,说明ERS参与INH致肝细胞损伤过程。但组蛋白乙酰化是否参与调控ERS,继而导致肝细胞损伤尚不清楚。
研究报道,在大鼠心肌缺血再灌注损伤中,曲古霉素A可上调ERS相关蛋白GRP78启动子处组蛋白H4乙酰化水平[22];丙戊酸可通过抑制HDAC活性提高GRP78蛋白表达和整体组蛋白H3的乙酰化水平,抑制CHOP上调,促进内质网分子伴侣表达,减轻ERS诱导的糖尿病肾病细胞凋亡[23]。组蛋白乙酰化可促进CFTR基因表达,诱发ERS,促进高同型半胱氨酸所致损伤肝细胞的凋亡[24]。上述研究表明,组蛋白乙酰化可在一定程度上调控ERS,但其调控ERS参与肝脏疾病的报道鲜见。组蛋白乙酰化酶抑制剂C646对P300具有高度选择性,可通过结合到P300的Lys-CoA结构区对其功能产生竞争性抑制;而组蛋白去乙酰化酶抑制剂MS-275能显著抑制HDAC1活性,增加细胞内组蛋白乙酰化程度。为进一步研究INH致肝细胞损伤中组蛋白乙酰化对ERS的调控作用,我们在制备肝细胞损伤模型同时分别加入C646或MS-275干预,结果显示,与INH组比较,INH+C646组中P300 mRNA表达和蛋白含量均明显降低,说明其组蛋白乙酰化水平降低,而ERS标志性分子GRP78 mRNA表达和蛋白含量均明显下降,CHOP mRNA表达和蛋白含量均明显升高,说明肝细胞损伤加重;与INH组比较,INH+MS-275组HDAC1 mRNA表达和蛋白含量均明显降低,说明组蛋白乙酰化水平升高,GRP78 mRNA表达和蛋白含量均明显升高,CHOP mRNA表达和蛋白含量均明显降低,说明ERS得到缓解,肝细胞损伤减轻。由此可见,在INH致肝细胞损伤过程中,C646和MS-275均可通过改变细胞内组蛋白乙酰化状态参与调控ERS,进而影响肝细胞损伤的发生、发展。
综上所述,组蛋白乙酰化通过调控ERS参与INH致肝细胞损伤。但组蛋白乙酰化参与ERS的具体作用机制尚不完全清楚,仍需进一步研究。
参考文献:
[1] Koul A, Arnoult E, Lounis N, et al. The challenge of new drug discovery for tuberculosis[J]. Nature, 2011,469(7331):483-490.
[2] Bjornsson ES, Bergmann OM, Bjornsson HK, et al. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland[J]. Gastroenterology, 2013,144(7):1419-1425.
[3] Sarich TC, Adams SP, Petricca G, et al. Inhibition of isoniazid-induced hepatotoxicity in rabbits by pretreatment with an amidase inhibitor[J]. J Pharmacol Exp Ther, 1999,289(2):695-702.
[4] Shu CC, Lee CH, Lee MC, et al. Hepatotoxicity due to first-line anti-tuberculosis drugs: a five-year experience in a Taiwan medical centre[J]. Int J Tuberc Lung Dis, 2013,17(7):934-939.
[5] Chun P. Histone deacetylase inhibitors in hematological malignancies and solid tumors[J]. Arch Pharm Res, 2015,38(6):933-949.
[6] Wang Y, Yang Y, Luo Y, et al. Aberrant histone modification in peripheral blood B cells from patients with systemic sclerosis[J]. Clin Immunol, 2013,149(1):46-54.
[7] Tao SC, Yuan T, Rui BY, et al. Exosomes derived from human platelet-rich plasma prevent apoptosis induced by glucocorticoid-associated endoplasmic reticulum stress in rat osteonecrosis of the femoral head via the Akt/Bad/Bcl-2 signal pathway[J]. Theranostics, 2017,7(3):733-750.
[8] Baumeister P, Luo S, Skarnes WC, et al. Endoplasmic reticulum stress induction of the Grp78/BiP promoter: activating mechanisms mediated by YY1 and its interactive chromatin modifiers[J]. Mol Cell Biol, 2005,25(11):4529-4540.
[9] Sinha-Hikim AP, Sinha-Hikim I, Friedman TC. Connection of nicotine to diet-induced obesity and non-alcoholic fatty liver disease: cellular and mechanistic insights[J]. Front Endocrinol (Lausanne), 2017(8):23.
[10] van den Boscha T, Boichenkob A, Leus NGJ, et al. The histone acetyltransferase p300 inhibitor C646 reduces proinflammatory gene expression and inhibits histone deacetylases[J]. Biochem Pharmacol, 2016(102):130-140.
[11] Bowers EM, Yan G, Mukherjee C, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase:identification of a selective small molecule inhibitor[J]. Chem Biol, 2010,17(5):471-482.
[12] Lin WH, Martin JL, Marsh DJ, et al. Involvement of insulin-like growth factor-binding protein-3 in the effects of histone deacetylase inhibitor MS-275 in hepatoma cells[J]. J Biol Chem, 2011,286(34):29540-29547.
[13] 牛琛,张一杨,李金凤,等.异烟肼对人肝细胞CYP1A1启动子区甲基化的影响[J].山东医药,2017,57(26):1-4.
[14] Gräff J, Tsai LH. Histone acetylation: molecular mnemonics on the chromatin[J]. Nat Rev Neurosci, 2013,14(2):97-111.
[15] 朱凌妍,李玉红,任琦,等.异烟肼致大鼠肝损伤使组蛋白H4低乙酰化[J].中国药理学与毒理学杂,2016,30(11):1192-1196.
[16] Tuzcu H, Unal B, Kirac E, et al. Neutral sphingomyelinase inhibition alleviates apoptosis, but not ER stress, in liver ischemia-reperfusion injury[J]. Free Radic Res, 2017,51(3):253-268.
[17] Henkel AS, LeCuyer B, Olivares S, et al. Endoplasmic reticulum stress regulates hepatic bile acid metabolism in mice[J]. Cell Mol Gastroenterol Hepatol, 2016,3(2):261-271.
[18] Bhoola NH, Kramvis A. Hepatitis B e antigen expression by hepatitis B virus subgenotype A1 relative to subgenotypes A2 and D3 in cultured hepatocellular carcinoma (Huh7) cells[J]. Intervirology, 2016,59(1):48-59.
[19] Takahara I, Akazawa Y, Tabuchi M, et al. Toyocamycin attenuates free fatty acid-induced hepatic steatosis and apoptosis in cultured hepatocytes and ameliorates nonalcoholic fatty liver disease in mice[J]. PLoS One, 2017,12(3):e0170591.
[20] He L, Hou X, Fan F, et al. Quercetin stimulates mitochondrial apoptosis dependent on activation of endoplasmic reticulum stress in hepatic stellate cells[J]. Pharm Biol, 2016,54(12):3237-3243.
[21] Afrin R, Arumugam S, Soetikno V, et al. Curcumin ameliorates streptozotocin-induced liver damage through modulation of endoplasmic reticulum stress-mediated apoptosis in diabetic rats[J]. Free Radic Res, 2015,49(3):279-289.
[22] Yu L, Lu M, Wang P, et al. Trichostatin A ameliorates myocardial ischemia/reperfusion injury through inhibition of endoplasmic reticulum stress-induced apoptosis[J]. Arch Med Res, 2012,43(3):190-196.
[23] Sun XY, Qin HJ, Zhang Z, et al. Valproate attenuates diabetic nephropathy through inhibition of endoplasmic reticulum stressinduced apoptosis[J]. Mol Med Rep, 2016,13(1):661-668.
[24] Yang A, Sun Y, Mao C, et al. Folate protects hepatocytes of hyperhomocysteinemia mice from apoptosis via cystic fibrosis transmembrane conductance regulator (CFTR)-activated endoplasmic reticulum stress[J]. J Cell Biochem, 2017,118(9):2921-2932.
