以臂丛神经损害为首发症状的遗传性压力易感性周围神经病一例临床表型及基因突变分析
2018-05-09刘晴陈海雷霖朱文佳邸丽卢岩王敏王锁彬笪宇威
刘晴 陈海 雷霖 朱文佳 邸丽 卢岩 王敏 王锁彬 笪宇威
遗传性压力易感性周围神经病(HNPP)是常染色体显性遗传性周围神经病,系PMP22基因突变所致,通常于青少年期起病,由轻微甚至难以察觉的压迫、牵拉诱发,表现为易嵌压部位复发性、无痛性单神经病或多神经病,病理学可见典型“腊肠”样改变。以臂丛神经损害为首发表现者临床少见,本文回顾分析1例以臂丛神经损害为首发症状的遗传性压力易感性周围神经病患者的临床表型和基因突变特点,以期提高临床医师对疾病的认识,减少误诊和漏诊。
病例资料
患者 男性,46岁,因左侧肩部不适感伴左上肢无力2月余,于2016年10月25日入院。患者2月余前吹空调后突然出现左侧肩部不适感,表现为肩部肿胀,无明显麻木、无力,症状进行性加重,未予特殊处理;3 d后突感开车时转方向盘费力,予颈肩部按摩治疗,4 d后出现左上肢明显无力,表现为左臂上举、外展、旋前不能,持物、写字尚可,同时出现肩部、左上臂外侧和左手拇指轻微麻木感,当地医院行肌电图检查(2016年9月1日),神经传导速度(NCV)显示,左侧胸前外侧神经和内侧神经、腋神经、肩胛上神经、桡神经运动神经传导波幅降低,双侧正中神经、胫神经运动神经传导潜伏期延长;双侧尺神经、正中神经、胫神经感觉神经传导速度减慢,双侧尺神经、桡神经、正中神经、胫神经感觉神经传导波幅降低,双侧正中神经感觉神经传导潜伏期延长,提示左上肢臂丛上干损害,四肢周围神经损害(感觉神经重于运动神经),髓鞘和轴索损伤。临床诊断为“臂丛神经炎”,予营养神经、改善循环等对症治疗(具体方案不详),左上肢无力症状逐渐好转,左臂旋前恢复,左手可搭至右侧肩部,麻木症状好转。为求进一步诊断与治疗遂至我院就诊。患者自发病以来,精神、睡眠、饮食可,大小便正常,体重无明显变化。
既往史、个人史及家族史 患者10余年前高处坠落伤致颅脑创伤,未遗留后遗症;耳鸣2年余,未予处理;吸烟20余年、10支/d,饮酒20余年,150 g/d;父母身体健康,非近亲婚配,无明确家族性遗传性疾病病史。
体格检查 血压110/70 mm Hg(1 mm Hg=0.133 kPa),神志清楚,语言流利,高级皮质功能正常,脑神经检查未见异常;左上臂肌萎缩,余肌容积正常,左上肢外展、旋前不能,左上肢近端肌力2级、远端5级,右上肢和双下肢肌力5级,四肢肌张力正常,双足无畸形,步态正常,双侧指鼻试验、快复轮替动作、跟⁃膝⁃胫试验稳准,深浅感觉正常,四肢腱反射降低,病理征阴性,脑膜刺激征阴性。
辅助检查 实验室检查:血尿便常规,血液生化,凝血功能,甲状腺功能试验,血清红细胞沉降率(ESR)、维生素B12(VB12),类风湿因子(RF)、抗溶血性链球菌素O(ASO)、C⁃反应蛋白(CRP),IgA、IgG、IgM和补体C3、C4,乙型肝炎病毒(HBV)、梅毒螺旋体(TP)、人类免疫缺陷病毒(HIV),肿瘤标志物,抗核抗体(ANA)谱、抗中性粒细胞胞质抗体(ANCA)谱、副肿瘤相关抗体、莱姆病抗体、布鲁菌病抗体、抗神经节苷脂GM1抗体均于正常值范围;腰椎穿刺脑脊液检查,外观清亮、透明,压力为170 mm H2O(1 mm H2O=9.81×10⁃3kPa,80~180 mm H2O),蛋白定量710 mg/L(150~450 mg/L)、葡萄糖和氯化物于正常值范围,寡克隆区带(OB)和特异性寡克隆区带(SOB)呈弱阳性,脑脊液涂片(新型隐球菌、抗酸染色、革兰染色)呈阴性,TORCH 10项[弓形虫(TOX)、风疹病毒(RV)、巨细胞病毒(CMV)、Ⅰ型和Ⅱ型单纯疱疹病毒(HSV⁃1和HSV⁃2)]、副肿瘤相关抗体、莱姆病抗体、布鲁菌病抗体、抗神经节苷脂GM1抗体均于正常值范围。影像学检查:头部MRI未见明确病灶,副鼻窦炎。颈椎MRI显示,颈椎退行性变,C6⁃7左侧椎间孔狭窄(图1)。臂丛超声显示,双侧臂丛神经对称,未见明显异常。腹部超声显示,左侧肾结石。胸部CT未见明确病灶。神经电生理学检查:心电图呈窦性心率。肌电图(2016年10月31日)显示,双侧尺神经、正中神经、胫神经运动神经传导速度减慢、潜伏期延长;双侧尺神经、正中神经感觉神经传导未引出肯定波形,双侧胫神经感觉神经传导速度减慢、波幅降低,提示四肢周围神经损害(运动神经和感觉神经均受累)。
基因检测 采集患者外周静脉血4 ml,送检北京金准基因科技有限责任公司,采用目标区域捕获高通量测序技术检测PMP22基因,结果显示,PMP22基因外显子区大片段杂合缺失突变(图2)。
诊断与治疗 临床诊断为臂丛神经损害(臂丛神经炎可能性大,不排除遗传性压力易感性周围神经病),予以前列地尔10 μg/d静脉滴注改善循环、七叶皂苷钠20 mg/d静脉滴注减轻神经水肿,以及甲钴胺1 mg/d、维生素B10.10 g/d静脉注射营养神经。患者住院13 d,出院后继续服用B族维生素营养神经治疗,出院后2个月随访,左上肢肌力基本恢复正常。追问家族史,诉患者之子、姊及其子均有相似临床症状,进一步对上述家族成员进行基因检测,证实为遗传性压力易感性周围神经病家系(图3)。
讨 论
患者中年男性,急性发病,以左侧臂丛上干损害为首发表现,无疼痛、麻木等不适感,无明确嵌压史和家族史;体格检查除左侧臂丛上干损害表现外,四肢腱反射降低;实验室和影像学检查均无特殊;神经电生理学检查提示四肢广泛性神经传导异常,感觉神经重于运动神经,髓鞘和轴索损伤;基因检测提示PMP22基因杂合缺失突变,明确诊断为遗传性压力易感性周围神经病。

图1 颈椎MRI检查所见 1a 矢状位T1WI显示,颈椎退行性变(箭头所示)1b 横断面T2WI显示,C6⁃7左侧椎间孔狭窄(箭头所示)Figure 1 Cervical MRI findings Sagittal T1WI showed cervical retrograde degeneration(arrow indicates,Panel 1a).AxialT2WI showed left inter vertebral for amen stenosis of C6-7(arrow indicates,Panel 1b).

图2 PMP22基因检测显示,PMP22基因重复突变导致腓骨肌萎缩症,PMP22基因杂合缺失突变导致遗传性压力易感性周围神经病Figure 2 PMP22 gene detection showed that repeated mutation of PMP22 gene resulted in CMT,and the loss of heterozygosity of PMP22 gene resulted in HNPP.
遗传性压力易感性周围神经病高峰发病年龄约30岁,亦有早发病例报道,男女比例大致相同。Meretoja等[1]的流行病学调查研究显示,芬兰西南部435 000人群遗传性压力易感性周围神经病患病率约为16/10万,低于PMP22基因重复突变导致的腓骨肌萎缩症1A型(CMT1A型),可能与遗传性压力易感性周围神经病患者症状轻微且可自行恢复有关。目前,国内尚无关于遗传性压力易感性周围神经病的流行病学调查。
遗传性压力易感性周围神经病临床主要表现为易嵌压部位复发性、无痛性单神经病或多神经病。典型患者通常有家族史,急性发病,于轻微压迫、牵拉后出现运动和(或)感觉障碍,感觉障碍均为非痛性。最易受累神经是易嵌压部位神经,一项纳入70例遗传性压力易感性周围神经病患者的临床研究显示,麻痹发作时受累神经分别为腓神经36%、尺神经28%、臂丛神经20%、桡神经13%[2]。急性期体格检查可见受累神经支配区域肌力下降和(或)感觉障碍,腱反射可正常、降低或消失,亦可见受累部位肌萎缩。临床症状与体征多于数周或数月内自行恢复,多次损伤同一神经可能遗留部分神经功能缺损。约10%的遗传性压力易感性周围神经病患者出现与腓骨肌萎缩症相似的高弓足。此外,遗传性压力易感性周围神经病患者还表现为不典型症状,如全身无力、痉挛、慢性尺神经病变、腕管综合征(CTS)、吉兰⁃巴雷综合征(GBS)等[3]。该例患者呈急性发病,为非痛性神经损害,体格检查可见肌萎缩、肌力下降和腱反射降低,无高弓足,发病后5个月自行恢复,但无明确嵌压史和家族史,有长期饮酒史,易误诊为酒精性周围神经病。
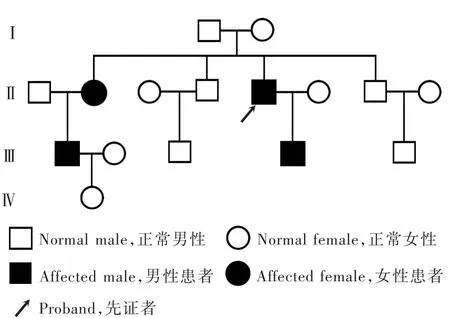
图3 遗传性压力易感性周围神经病家系图Figure 3 Family diagram of HNPP.
遗传性压力易感性周围神经病的神经电生理学检查十分重要,此类患者神经系统症状较轻微甚至无症状,易误诊或漏诊。无论临床是否有神经系统症状与体征,神经电生理学检查均可见弥漫性和广泛性神经传导异常,运动神经和感觉神经均受累,主要为脱髓鞘改变,即运动神经传导速度减慢、潜伏期延长,嵌压部位神经传导阻滞,F波潜伏期延长甚至消失。1999年,Mouton等[2]对99例染色体17p11.2缺失的遗传性压力易感性周围神经病患者的临床表现和神经电生理学进行研究,结果显示,尽管临床表现各异,但神经电生理学检查是可靠、经济的筛查工具,并将双侧正中神经感觉和运动神经传导速度减慢、单侧腓总神经传导远端潜伏期延长作为诊断标准。Tackenberg等[4]的研究显示,遗传性压力易感性周围神经病患者脑干听觉诱发电位(BAEP)、瞬目反射和咀嚼反射等潜伏期均较正常对照者延长,表明此类患者脑神经和中枢神经均受累。另一项纳入46例遗传性压力易感性周围神经病患者的神经电生理学研究显示,39例患者(84.78%)存在腕关节远端运动神经传导潜伏期延长[5]。本文患者肌电图可见四肢广泛性神经传导异常,感觉神经重于运动神经,符合遗传性压力易感性周围神经病的诊断标准,但临床应注意与腓骨肌萎缩症和酒精性周围神经病相鉴别。
遗传性压力易感性周围神经病的主要病理改变是局灶性增厚的髓鞘之间存在正常髓鞘区域,沿神经纤维纵轴形似“腊肠”,故又称“腊肠样神经病”。1972 年,Behse等[6]描述“腊肠”样结构并非遗传性压力易感性周围神经病的特征性病理改变,如CMT1B型、CMT3型、IgM副蛋白血症神经病和慢性炎性脱髓鞘性多发性神经根神经病(CIDP)等[7]亦可见“腊肠”样结构。
迄今发现的与遗传性压力易感性周围神经病关系最密切的基因是PMP22基因,约80%系位于染色体17p11.2的长度为1.50×103bp的片段缺失所致,亦有部分系PMP22基因重复突变所致[8]。正常人群存在2个拷贝的PMP22基因,若在减数分裂过程中发生不均等交换则仅使1个PMP22基因突变,导致遗传性压力易感性周围神经病,亦可发生3个PMP22基因突变,导致CMT1A型[9]。本文患者基因检测显示,PMP22基因外显子区杂合缺失突变。
遗传性压力易感性周围神经病的诊断要点包括[10]:(1)临床出现反复发作性单神经病或多神经病。(2)神经电生理学表现为广泛性神经传导异常,主要为脱髓鞘改变。(3)周围神经组织病理学可见“腊肠”样结构。(4)阳性家族史。(5)基因检测显示染色体17p11.2大片段缺失。应注意与以下疾病相鉴别:(1)遗传性神经痛性肌萎缩(HNA),为常染色体显性遗传性疾病,通常于10~20岁发病,临床主要表现为发作性痛性臂丛神经损害,肌萎缩较明显,因此,应注意与单纯表现为臂丛神经损害的遗传性压力易感性周围神经病相鉴别。肌电图显示远端神经传导基本正常,无类似遗传性压力易感性周围神经病的广泛性神经传导异常。(2)炎性脱髓鞘性多发性神经根神经病,好发于青年男性,是一种自身免疫性疾病,分为急性和慢性两种类型,脑脊液可见蛋白⁃细胞分离现象,通常无家族史,激素、静脉注射免疫球蛋白、免疫抑制剂治疗有效,可资鉴别。本文患者发病前有吹空调受凉史,否认嵌压史和家族史,易诊断为臂丛神经炎,四肢腱反射降低曾考虑与长期大量饮酒有关,但为无痛性神经损害,神经电生理学可见广泛性神经传导异常,可资鉴别。
目前,遗传性压力易感性周围神经病的治疗主要是营养神经和对症支持治疗,急性期不主张物理康复治疗,神经损害通常于几周内恢复,若出现疼痛,可予抗抑郁药、抗癫药和细胞膜稳定剂等。因此,遗传性压力易感性周围神经病患者关键在于预防,避免神经牵拉和局部压迫等诱因,减少神经损害,避免不恰当治疗(如外科手术)。仅少数反复同一部位复发患者可能遗留神经功能缺损,大部分患者预后良好。本文患者服用营养神经药并进行肢体康复锻炼,发病后5个月(出院后2个月)随访,左上肢肌力基本正常。
综上所述,本文患者是以单纯臂丛上干损害为首发症状的遗传性压力易感性周围神经病,无明确嵌压史和家族史,极易误诊和漏诊,提示临床实践中如果遇到表现为臂丛神经损害而神经电生理学提示广泛性神经传导异常的患者,应高度警惕遗传性压力易感性周围神经病,以提高疾病诊断率并减少神经损害发生率。
[1]Meretoja P,Silander K,Kalimo H,Aula P,Meretoja A,Savontaus ML. Epidemiology of hereditary neuropathy with liability to pressure palsies(HNPP)in south western Finland[J].Neuromuscul Disord,1997,7:529⁃532.
[2]Mouton P,Tanlieu S,Goulder R,Birouk N,Maisonobe T,Dubourg O,Brice A,LeGuern E,Bouche P.Spectrum of clinical and electrophysiologic features in HNPP patients with the 17p11.2 deletion[J].Neurology,1999,52:1440⁃1446.
[3]Luigetti M,Del Grande A,Conte A,Lo Monaco M,Bisogni G,Romano A,Zollino M,Rossini PM,Sabatelli M.Clinical,neurophysiological and pathological findings of HNPP patients with 17p12 deletion:a single⁃centre experience[J].J Neurol Sci,2014,341(1/2):46⁃50.
[4]Tackenberg B,Möller JC,Rindock H,Bien S,Sommer N,Oertel WH,Rosenow F,Schepelmann K,Hamer HM,Bandmann O.CNS involvement in hereditary neuropathy with pressure palsies(HNPP[)J].Neurology,2006,67:2250⁃2252.
[5]Sacbiko T,Marvin C,Kurt K.Electrodiagnostic characterization of hereditary neuropathy with liability to pressure palsies[J].J Clin Neuromuscul Dis,2017,18:119⁃124.
[6]Behse F,Bucbthal F,Carlsen F,Knappeis GG.Hereditary neuropathy with liability to pressure palsies:electrophysiological and histopathologicalaspects[J].Brain,1972,95:777⁃794.
[7]Sander S,Ouvrier RA,Mcleod JG,Nicholson GA,Pollard JD.Clinicalsyndromes associated with to tomacula ormyelin swellings in suralnerve biopsies[J].J Neurol Neurosurg Psychiatry,2000,68:483⁃488.
[8]NelisE,Van Broeckhoven C,De Jonghe P,Löfgren A,Vandenberghe A,Latour P,Le Guern E,Brice A,Mostacciuolo ML,Schiavon F,Palau F,Bort S,Upadhyaya M,Rocchi M,Archidiacono N,Mandich P,Bellone E,Silander K,Savontaus ML,Navon R,Goldberg⁃Stern H,Estivill X,Volpini V,Friedl W,Gal A.Estimation of the mutation frequencies in Charcot⁃Marie⁃Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies:a European collaborative study[J].Eur J Hum Genet,1996,4:25⁃33.
[9]Lupski JR.Charcot⁃Marie⁃Tooth disease:a gene⁃dosage effect[J].Hosp Pract,1997,32:83⁃84.
[10]CuiF,Huang XS.Advances in the study of hereditary neuropathy with liability to pressure palsies[J].Di San Jun Yi Da Xue Xue Bao,2004,26:647⁃648[.崔芳,黄旭升.遗传性压迫易感性神经病的研究进展[J].第三军医大学学报,2004,26:647⁃648.]
