Mo掺杂W18O49复合材料作为氧还原催化剂载体的研究
2018-05-08,,,
, , ,
(浙江工业大学 化学工程学院,浙江 杭州 310014)

在本研究工作中,笔者尝试在W18O49中掺杂不同比例的Mo原子,制备系列Mo—W18O49复合材料,并从中确定W/Mo的最优原子比,再在Mo—W18O49上负载少量的Pt纳米粒子(质量分数10%).通过电化学测试发现,相比于Pt/W18O49和商用的20% Pt/C,Pt/30% Mo—W18O49电催化剂在酸性介质中不仅表现出更为优异的氧还原催化性能,而且还有较好的稳定性.这种独特的催化活性能够归因于在W18O49中掺杂了Mo元素而引入的丰富的氧缺陷以及更强的化学相互作用和电子耦合效应.
1 实验部分
1.1 氧化钨(W18O49)电催化剂的合成
称取一定量的WCl6(0.05 g)溶解到20 mL的乙醇溶剂中,搅拌10 min至WCl6完全溶解,将所得到的均一溶液转移至50 mL的水热反应釜中,200 ℃条件下保持4 h,冷却到室温后,将生成的沉淀物用无水乙醇和去离子水离心洗涤3次,以去除杂质离子,最后在60 ℃真空条件下干燥过夜,得到蓝色的W18O49粉末备用.
1.2 Mo掺杂W18O49载体的制备
称取适量摩尔比的WCl6和MoCl5,将其分别溶于10 mL的乙醇中,搅拌10 min,再将两管溶液混合,继续搅拌5 min,然后将得到的均一混合溶液转移至50 mL的水热反应釜中,200 ℃条件下保持4 h,冷却到室温后,将生成的沉淀物用无水乙醇和去离子水离心洗涤3次,以去除杂质离子,最后在60 ℃真空条件下干燥过夜,得到黑色粉末.对于掺杂不同W与Mo摩尔比的样品分别标记为W18O49(0 Mo),10%Mo—W18O49,30% Mo—W18O49,50% Mo—W18O49.
1.3 Pt/W18O49和Pt/30% Mo—W18O49电催化剂的制备
W18O49和30% Mo—W18O49的载Pt量均为质量分数10%,采用微波辅助多元醇法载Pt.将适量的H2PtCl6(5 mmol/L)分别加入到已制备好的W18O49和30% Mo—W18O49中,超声30 min,使W18O49和30% Mo—W18O49能够均匀地分散到H2PtCl6溶液中,然后向上述悬浊液中分别加入10 mL的乙二醇,搅拌5 min使其混合均匀,再用适量的KOH溶液将混合溶液的pH调到8~10,最后将混合溶液在微波辅助加热条件下,180 ℃反应30 min,将生成的产物用去离子水和无水乙醇离心洗涤3次,最后在真空条件下60 ℃,干燥过夜,备用.
1.4 催化剂结构和形貌的表征
材料的晶体结构用X射线衍射分析(XRD),XRD图谱采用荷兰PANalytical(帕纳科)公司生产的X’Pert PRO型X射线衍射仪分析.X射线源为Cu靶 Kα射线(λ=0.154 056 nm),电压40 kV,电流40 mA,扫描范围为10°~80°.TEM图片由JEM-2010Ex型高分辨透射电子显微镜(日本JEOL公司)测得,操作电压为200 kV.X射线能量色散光谱仪(EDX)光源采用Cu靶Kα射线(λ=0.154 056 nm),材料表面的元素及化合态采用KRATOS AXIS ULTRA型号的X射线光谱(XPS)分析;材料的分子结构采用法国JOBIN YVON公司的Lab RAM HR UV800型号激光拉曼光谱仪测定.
1.5 催化剂对氧还原电催化性能的测试
所有的电化学测试都采用上海辰华的CHI660C型号电化学工作站,在标准的三电极体系中进行,铂电极为对电极,饱和甘汞电极为参比电极,5 mm的旋转圆盘电极(RDE)为工作电极.所有的电极电位采用可逆氢电极(RHE)校正,即
E1=E2+0.059x+E3
式中:E1为可逆氢电极;E2为饱和甘汞电极;E3为饱和甘汞电极的标准电极电势;x为电解液的pH值.
在进行氧还原性能测试之前,先在N2饱和的0.5 mol/L的H2SO4溶液中进行循环伏安测试以活化电极,并去除溶液中的溶解氧气,电位扫描速率为50 mV/s,扫描范围为0.1~1.0 V,扫描圈数20圈;然后通氧气鼓泡30 min,使溶液中氧气达到饱和状态,在继续通氧气条件下以同样方法进行循环伏安测试,然后进行线性扫描测试,电位扫描速率为5 mV/s,扫描范围为1.0~0.1 V,转速分别为1 600,800,400,200,100 r/min.各个电极电位下的Koutecky-Levich曲线可以由下列等式得到,通过拟合出来的线性斜率,可以计算得到氧还原过程中的转移电子数n,即
B=0.62nFAD2/3V1/6CO2
式中:J为实验中测得的电流;JL为测得的极限扩散电流;JK为动力学电流;n为转移电子数;F为法拉第常数(96 485 C/mol);A为电极的几何面积(0.196 cm2);D为O2在0.5 mol/L的H2SO4中的扩散系数(1.4×10-5cm2/s);v为电解液的动力学黏度(0.01 cm2/s);ω为旋转角速度,ω=2πf/60,f为旋转圆盘电极的转速;CO2为O2在0.5 mol/L H2SO4中的浓度(1.1×10-5mol/L).
比活性是通过将计算得到的动力学电流JK归一化到贵金属Pt的电化学活性表面积(ECSA)得到的.在计算ECSA时,制备的电极要先在N2饱和的0.5 mol/L的H2SO4中进行循环伏安测试,电位扫描速率为50 mV/s,扫描范围为0.1~1.0 V,直到获得稳定的氢气吸脱附曲线.催化剂的稳定性测试是在O2饱和的0.5 mol/L的H2SO4溶液中通过在0.1~0.8 V范围内不断地进行循环伏安扫描(500圈)得到,扫描速率50 mV/s.
2 结果与讨论
2.1 催化剂的结构表征
图1为所制备的不同样品的XRD图谱,从图1中可以看出:无Mo掺杂的W18O19样品在23.33°和47.87°有2个明显的衍射峰,分别对应于单斜晶型W18O19的(010)和(020)晶面(JCPDS No: 03-065-1291),这和之前文献中报道相符合[14-15].而在有Mo掺杂的3个样品中,衍射峰的峰型与纯W18O19的峰型相比没有变化,仅仅是随着Mo掺杂量的增加,衍射峰的强度有所下降,尤其当Mo的掺杂量为50%时,位于23.3°附近的(010)晶面的衍射峰明显降低,而47.8°附近的(020)晶面的衍射峰几乎看不到.同时可以观察到随着Mo的掺杂量的增加,衍射峰的宽度增加了,可能是由于Mo的掺杂导致了所制备的Mo—W18O19载体的结晶度降低.从图1还可看到:与纯W18O19相比,随着Mo掺杂量的增加,所制备的Mo—W18O19的(020)处的峰位置依次向右偏移,这可能是由于Mo的有效离子(0.059 nm)半径比W(0.061 nm)要稍小,当Mo被引入到氧化钨的晶格当中时,引起氧化钨主体晶格间距变小,根据布拉格方程可知,衍射角度会相应变大.另外,在有Mo掺杂的3个样品中,没有任何与Mo相关的衍射峰出现,说明没有Mo相关的单独的晶相出现,证明了Mo成功掺杂到氧化钨的晶格当中,替换了部分W原子,同时仍保持W18O49的晶格结构.

图1 不同样品的XRD图谱Fig.1 XRD patterns of different samples
图2为纯W18O49、不同比例的Mo—W18O49载体以及Pt/30% Mo—W18O49样品的TEM图谱以及对应的HRTEM图谱.从图2(a~d)中可看出:当Mo的掺杂量不同时,所得到的产物的形貌并不相同.当未掺杂Mo原子时(图2a),产物呈现出长约为200 nm,宽约10 nm的一维纳米线状,根据之前的文献报道,当WCl6在乙醇中的质量浓度大于15 g/L时,所得到的氧化钨为海胆状;当质量浓度小于5 g/L时,产物形貌为纳米线状,由此本实验所得到的一维纳米线状的氧化钨与文献中相符合[14];图2(b)为当Mo的掺杂量为10%时,所制备出的催化剂载体,从中不难看出,尽管WCl6在乙醇中的质量浓度仍为2.5 g/L,因为有10%的MoCl5的加入,使得溶液中盐浓度增加,从而呈现出图2(a)的纳米线相互聚集而成为簇状结构的趋势;当Mo的掺杂量继续增加到30%时(图2c),由于溶液中的盐浓度进一步增大,使得之前的线状结构进一步聚集,几乎呈现出海胆状,该形貌虽然是从线状结构演变而来,但是能明显看出其触须比海胆状更长并且更尖锐,同时从中间颜色上的深浅不难看出,尽管类似海胆状形貌,但中间球形部分更为疏松;图2(d)为Mo的掺杂量上升至50%时所得产物的形貌图,由于体系中盐质量浓度已几乎接近于文献中所给的临界值,并且2种金属阳离子之间相互影响,使得产物形貌完全失去图2(a)中的线状结构,呈现出带有较短触须的球状结构.从图2(f~i)中可看出:未掺杂Mo原子时,所得的产物晶格间距d=0.380 nm,对应于W18O49的(010)晶面,而随着Mo的掺杂量的增大,所得产物的晶格间距依次稍微减小,并且结晶度明显变差,正如之前所述,Mo的原子半径稍微小于W的原子半径,当Mo原子被掺杂进氧化钨的晶格当中时,便会引起氧化钨主体晶格间距的变小,这与之前XRD中所观察到的衍射峰的偏移现象相一致,也再次证明了Mo被成功掺杂进氧化钨的晶格当中.图2(e,j)为Pt/30% Mo—W18O49样品上Pt颗粒的TEM图,可看出Pt颗粒均匀的分布在30%Mo—W18O49载体上,颗粒尺寸为3~5 nm,测得的d=0.227 nm,对应于Pt的(111)晶面.

图2 不同样品的TEM图谱(上)以及对应的HRTEM图谱(下)Fig 2 The TEM images of different samples (above) and the corresponding HRTEM images (below)
X射线光电子能谱(XPS)能够被用来探测材料表面的化学组成以及状态.如图3(a)所示,30% Mo—W18O49的全谱显示了Mo,W,O以及一些由于污染所产生的C元素的存在.图3(b)能够被分成2对裂分峰,分别归属于W的2个不同的氧化态,从图3(b)中拟合后的线能够看出,位于38.06 eV和35.95 eV处的W 4f5/2和W 4f7/2峰归属于W6+,而在更低结合能处的37.5 eV和35.3 eV的W 4f5/2和W 4f7/2峰则对应于W5+.与文献中报道的纯W18O49相比,这3个典型的氧化态均明显向更高的结合能偏移,这可能是由于W6+(W5+)周围Mo原子的存在所导致[16-17].另外,XPS的半定量分析显示W5+与W6+的比例为0.52,说明了由于Mo的掺杂导致了更高浓度的氧缺陷的存在.从图3(c)中能够看出:Mo的3d谱能够分出3对裂分峰,第1对是位于236.09 eV的Mo 3d3/2和232.95 eV的Mo 3d5/2,归属于Mo6+;第2对是位于234.86 eV的Mo 3d3/2和231.7 eV的Mo 3d5/2,对应于Mo5+;第3对裂分峰则是位于234.86 eV的Mo 3d3/2和231.7 eV的Mo 3d5/2,来源于Mo4+[16,18-19].从图3(c)中Mo的3个价态的相对强度可看出:具有较高含量+5的Mo,正是由于Mo被掺杂到氧化钨中以后,产生了大量的氧缺陷所致,增加了材料的导电性的同时,能够促进O—O键的断裂,加速氧还原反应;另外,W和Mo所含的多种混合价态(+4,+5,+6),可以为O2的吸附提供化学吸附位点,并且能够极大地促进材料中电子的流动和传输[20-21].同时考虑到W和Mo原子电负性的不同,电子会从Mo原子(2.16)上转移到W原子(2.36)上,使W上带有更多的负电荷,而Mo上则相对带有更多的正电荷,有利于O2的吸附,这也就使得Mo能够成为氧还原催化剂中的另一个活性位点[22-23];当W上带有更多的负电荷以后,会进一步调节其上负载的Pt的电子结构,进而弱化Pt对于氧还原过程中产生的含氧物种(OH-)的吸附,使其及时脱出,留下干净的Pt表面,利于O2的进一步吸附和解离.图3(d)为O 1s的XPS分峰谱图,图中O的1s谱能够分出3个裂分峰,位于530.63 eV结合能处的裂分峰对应于晶格中的O原子(O2-),位于532.5 eV结合能处的裂分峰则归属于吸附的空气中的水分子或碳氧化物[16,24-25].对于出现在531.5 eV结合能处的裂分峰则是来源于掺杂所产生的氧缺陷和吸附的OH-,对于材料中的氧缺陷如前面所述,能够为O2的吸附提供活性位点,活化O—O键,促进其断裂;材料对OH-有一定的吸附能力,则说明如果Pt上吸附了氧还原过程中产生的中间含氧物种(OH-),载体中的O原子能够帮助其转移一部分的OH-到载体上,从而使负载的Pt能够持续保持较高的催化活性[25].
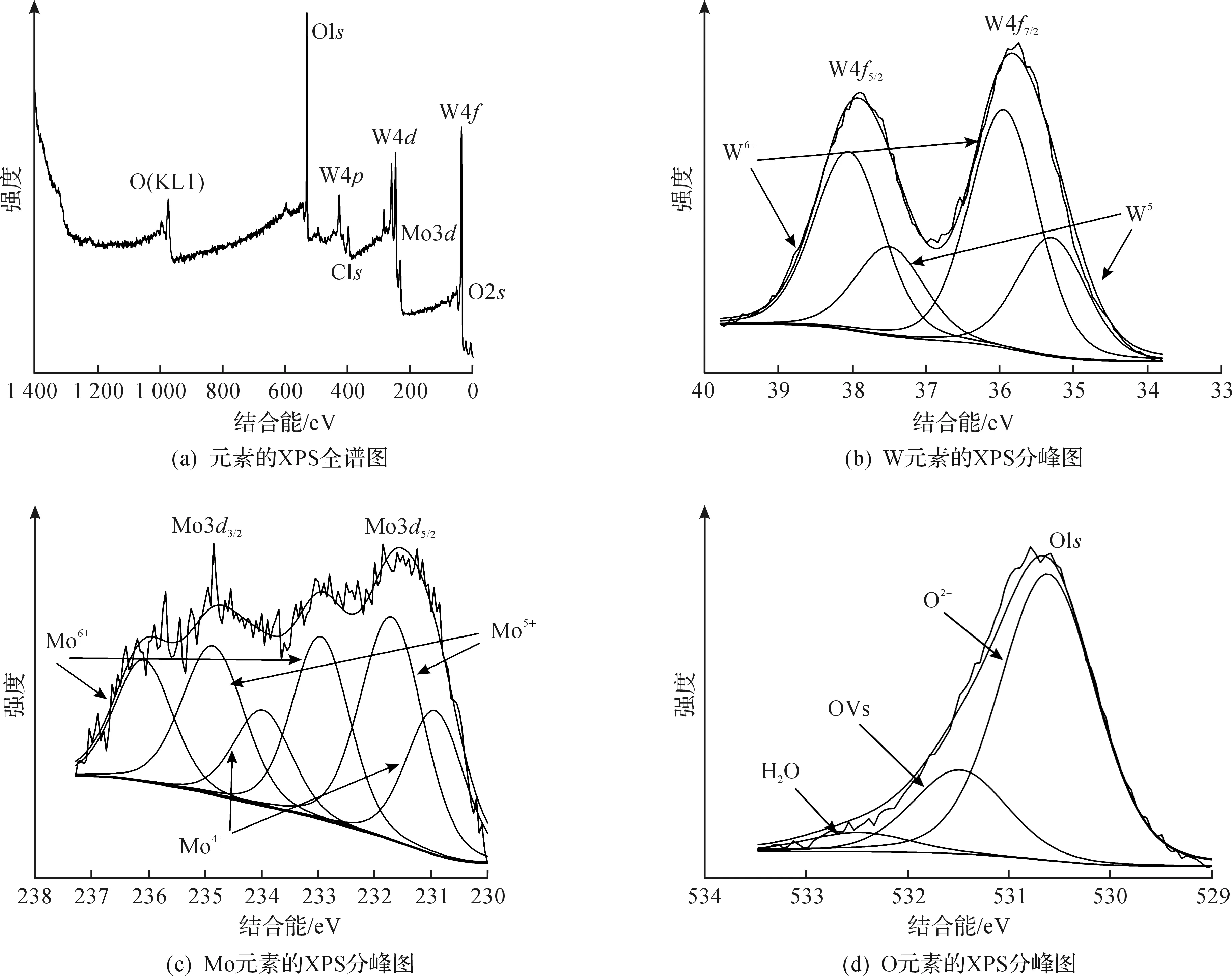
图3 30% Mo—W18O49电催化剂的XPS谱图Fig 3 The XPS spectra of the 30% Mo—W18O49 electrocatalyst
2.2 催化剂的电化学测试
为了研究不同W与Mo摩尔比的载体材料对氧还原催化性能的影响,W18O49和MoOx以及掺杂不同比例的Mo—W18O49在O2饱和的0.5 mol/L的H2SO4溶液中的极化曲线如图4所示,旋转速率1 600 r/min,扫描速率5 mV/s,可以看出30%Mo—W18O49样品展现出最好的氧还原催化性能.经计算得到,相比于50% Mo—W18O49,10% Mo—W18O49,W18O49(0 Mo)以及MoOx(100% Mo)4个样品,起始电位分别正移302,289,213,377 mV.30% Mo—W18O49样品表现出更为优异的催化性能的原因可能是由于当Mo的掺杂量为30%时,产生的氧缺陷数量相比于掺杂量为10%时所制备的材料数量更多,并且W—Mo的协同效应更明显,而当Mo的掺杂量为50%时,作为前驱体的W18O49的结构已被破坏,材料的结晶度也明显下降,削弱了两者之间的协同作用.

图4 不同载体材料的氧还原极化曲线Fig.4 ORR polarization curves on different support materials
以30% Mo—W18O49和W18O49作为载体,在其上分别负载少量的贵金属Pt(质量分数10%),所得产物分别标记为Pt/30% Mo—W18O49和Pt/W18O49,将其作为氧还原的电催化剂,与商用20% Pt/C相比较,在O2饱和的0.5 mol/L的H2SO4中的氧还原极化曲线,旋转速率1 600 r/min,扫描速率5 mV/s,如图5所示.从图5可看出:将3个催化剂的极限电流均归一化到电极的几何面积以后,样品Pt/30% Mo—W18O49的极限电流相比于另外2个略大一些,但都接近于文献中所报道的理论值(约为6.0 mA/cm2)[26].另外,氧还原的起始电位Eon和半波电位E1/2能够被用来快速评判催化剂的电催化性能[27],从这2个角度分析,Pt/30% Mo—W18O49电催化剂的氧还原起始电位为0.905 V,半波电位为0.815 V,比Pt/W18O49(0.865 V,0.783 V)和商用20% Pt/C(0.816 V,0.727 V)均有明显的正移.这也说明了30%Mo—W18O49作为载体,同时也作为活性位点,在酸性介质中对氧还原有相对更高的催化活性.
图6为Pt/30% Mo—W18O49,Pt/W18O49,和商用20% Pt/C在不同电位下的K—L曲线,从3张图中能够看出:所有的曲线均有较好的线性相关性,每一张图中的直线也都有相似的斜率,说明在所测试的电极电位范围内,对于氧还原反应来说有相同的电子转移数,对于Pt/30% Mo—W18O49催化剂来说,选取的电极电位范围为0.9~0.825 V,经计算,拟合后直线的斜率约为1.62,转移电子数为3.96,而对于Pt/W18O49样品,直线的斜率和转移电子数分别为1.73和3.75,对于商用20% Pt/C来说,直线的斜率和转移电子数分别为2.01和3.68,通过对比可发现,Pt/30%Mo—W18O49催化剂对于氧还原的催化反应过程以4e-为主.
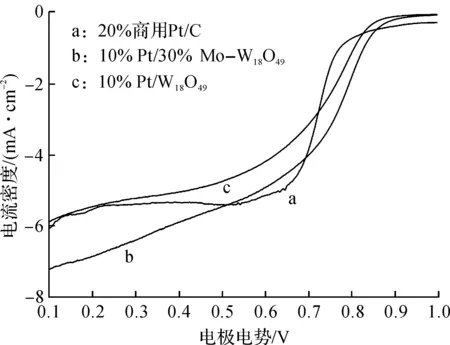
图5 不同催化剂的氧还原极化曲线Fig.5 ORR polarization curves on different samples

图6 不同催化剂的K—L曲线Fig.6 Koutecky levich plots on different samples
为了比较催化剂的本征活性,将Pt/30% Mo—W18O49,Pt/W18O49和商用20% Pt/C催化剂的动力学电流标准化到基于电化学活性表面积(ECSA)的比活性,催化剂的电化学活性表面积(ECSA)的计算公式可参考文献[28].如图7所示,通过计算系列Pt/Mo—W18O49催化剂的塔菲儿曲线斜率发现,3条曲线的斜率在整个电位区间内几乎都在-115 mV/dec左右,而根据文献中报道的,通常典型的塔菲儿曲线应该是在低过电位区曲线斜率为-60 mV/dec,高过电位区曲线斜率为-120 mV/dec,这2个斜率的产生与Pt表面氧化物的吸附和形成有关,主要是PtOH和PtO,它们均来源于Pt与H2O的反应,即

这些氧化物阻碍了氧还原反应的进行,从而直接影响到燃料电池的性能.而制备得到的Pt/Mo—W18O49系列催化剂尽管Mo的掺杂量不同,在整个电极电位区间内具有单一的较高的塔菲儿斜率,尤其是对于Pt/30% Mo—W18O49催化剂来说,在同样是0.9 V电位时,其有更大的动力学比活性,这是评价氧还原催化剂催化性能的一个直接标准,说明以30% Mo掺杂的W18O49作为载体材料,相比于其他2个催化剂有更强的能力,将Pt—OH的形成推向更高的电位,从而留下一个洁净的、高活性的Pt来促进氧还原反应[17].

图7 不同催化剂的塔菲儿曲线Fig.7 Tafel plots of different samples
图8为Pt/30% Mo—W18O49和20%商用Pt/C在0.5 mol/L的H2SO4中,扫描500圈循环伏安前后的极化曲线的对比图,扫描范围是0~1.0 V,转速为1 600 r/min.从图8中可以看出:循环伏安测试中(扫描范围0~1.0 V),在扫描500圈以后,图8(a)中Pt/30% Mo—W18O49催化剂的起始电位几乎没有变化,半波电位也仅仅负移15 mV左右,而在同样测试条件下图8(b)中的商用Pt/C的起始电位负移了68 mV,半波电位负移了约79 mV,再一次证明了由于Pt与30% Mo—W18O49载体间的强相互作用,抑制了反应过程中Pt颗粒的脱落以及及时去除氧气还原反应过程中吸附的含氧物种,从而使催化剂保持良好的稳定性.

图8 不同催化剂的稳定性对比图Fig.8 The comparison of stability of the different electrocatalyst
对于所合成的30% Mo—W18O49载体材料以及其对应的Pt/30% Mo—W18O49所展现出的更为优异的氧还原催化性能,首先,材料中存在的丰富的氧缺陷是增强氧还原催化性能的一个重要的因素.Mo同W一样,其自身的氧化物也带有较为丰富的氧缺陷,当Mo原子以合适的比例掺杂进W18O49晶格当中替换掉部分W原子的同时,在不破坏W18O49主体晶相的前提下,由于Mo的化合价更低,所需要与之平衡的O原子数目更少,自然会带出一部分O原子以保持整体的电中性,从而产生氧缺陷[29].从之前对XPS的分析中也能看出,W5+与W6+的比例为0.52,说明材料中含有大量的氧缺陷.而氧缺陷对于氧气还原的催化机理正如文献中所述,氧缺陷作为活性位点能够吸附O2分子,并将O—O键活化、拉长,最终导致其断裂,从而加速氧还原反应的动力学过程[12-13,29].
3 结 论
实验探究了不同比例Mo掺杂对所制备的Mo—W18O49材料形貌结构以及催化性能的影响,从中确定了30%为Mo的最优掺杂量.在所合成的30% Mo—W18O49载体上负载少量的Pt,并以其作为氧还原催化剂进行了电化学测试,发现所制备的Pt/30% Mo—W18O49电催化剂相比于商用Pt/C对于氧气还原反应有更好的催化性能.这种独特的催化活性一方面是由于Mo的掺杂使材料引入了大量的氧缺陷,这些氧缺陷能够作为活性中心吸附O2分子,加速催化O—O键的断裂;另一方面是归因于Pt与30% Mo—W18O49载体间存在的较强的相互作用,提高了材料的催化性能.
参考文献:
[1] 陈金媛,徐圣辰.杭州市PM2.5中PAEs污染现状与特征分析[J].浙江工业大学学报,2015,43(6):600-606.
[2] 蒋旭光,常威.生活垃圾焚烧飞灰的处置及应用概况[J].浙江工业大学学报,2015,43(1):7-17.
[3] 王珍.中国稀土资源开发和环境保护问题研究[J].浙江工业大学学报,2015,14(1):13-17.
[4] GRAY H B. Powering the planet with solar fuel[J]. Nature chemistry,2009,1(1):7.
采用Box-Behnken响应面分析法对艾纳香艾渣资源中总黄酮提取率工艺进行优化,得到艾渣提取最佳工艺参数为料液比1∶62(g∶mL),乙醇体积分数75%,提取时间31 min,超声功率400 W,艾渣总黄酮平均提取率为4.62%,验证试验结果与预测值相近,说明提取工艺稳定可行。在该工艺条件下,艾渣总黄酮的提取更为完全,为艾渣中总黄酮有效成分的回收利用提供参考,同时也为艾纳香废渣资源化开发利用提供技术依据。
[5] ZHANG G, SHEN S, GUO L, et al. Dynamic characteristics of local current densities and temperatures in proton exchange membrane fuel cells during reactant starvations[J]. International journal of hydrogen energy,2012,37(2):1884-1892.
[6] SHRESTHA S, LIU Y, MUSTAIN W E. Electrocatalytic activity and stability of pt clusters on state-of-the-art supports: a review[J]. Catalysis reviews,2011,53(3):256-336.
[7] YEO K M, CHOI S, ANISUR R M, et al. Surfactant-free platinum-on-gold nanodendrites with enhanced catalytic performance for oxygen reduction[J]. Angewandte chemie international edition,2011,50(3):745-748.
[8] SHIM J, LEE C R, LEE H K, et al. Electrochemical characteristics of Pt-WO3/C and Pt-TiO2/C electrocatalysts in a polymer electrolyte fuel cell[J]. Journal of power sources,2001,102(1/2):172-177.
[9] LEWERA A, TIMPERMAN L, ROGUSKA A, et al. Metal-support interactions between nanosized pt and metal oxides (WO3and TiO2) studied using X-ray photoelectron spectroscopy[J]. The Journal of physical chemistry C,2011,115(41):20153-20159.
[10] LI Q, WANG K, ZHANG S, et al. Effect of photocatalytic activity of CO oxidation on Pt/TiO2by strong interaction between Pt and TiO2under oxidizing atmosphere[J]. Journal of molecular catalysis A: chemical,2006,258(1/2):83-88.
[11] TSAI M C, NGUYEN T T, AKALEWORK N G, et al. Interplay between molybdenum dopant and oxygen vacancies in a TiO2support enhances the oxygen reduction reaction[J]. ACS catalysis,2016,6(10):6551-6559.
[12] JANG M H, AGARWAL R, NUKALA P, et al. Observing oxygen vacancy driven electroforming in Pt-TiO2-Pt device via strong metal support interaction[J]. Nano letters,2016,16(4):2139-2144.
[13] ZHOU H, SHI Y, DONG Q, et al. Surface oxygen vacancy-dependent electrocatalytic activity of W18O49nanowires[J]. The journal of physical chemistry C,2014,118(35):20100-20106.
[14] XI G, OUYANG S, LI P, et al. Ultrathin W18O49nanowires with diameters below 1 nm: synthesis, near-infrared absorption, photoluminescence, and photochemical reduction of carbon dioxide[J]. Angewandte chemie international edition,2012,51(10):2395-2399.
[15] BAI H, SU N, LI W, et al. W18O49nanowire networks for catalyzed dehydration of isopropyl alcohol to propylene under visible light[J]. Journal of materials chemistry A,2013,1(20):6125-6129.
[16] ZHONG X, SUN Y, CHEN X, et al. Mo doping induced more active sites in urchin-like W18O49nanostructure with remarkably enhanced performance for hydrogen evolution reaction[J]. Advanced functional materials,2016,26(32):5778-5786.
[17] LIU Y, SHRESTHA S, MUSTAIN W E. Synthesis of nanosize tungsten oxide and its evaluation as an electrocatalyst support for oxygen reduction in acid media[J]. ACS catalysis,2012,2(3):456-463.
[18] ESFAHANI R A M, VANKOVA S K, VIDELA A H A M,
et al. Innovative carbon-free low content Pt catalyst supported on Mo-doped titanium suboxide (Ti3O5-Mo) for stable and durable oxygen reduction reaction[J]. Applied catalysis B environmental,2017,201:419-429.
[20] CHENG F, SHEN J, PENG B, et al. Rapid room-temperature synthesis of nanocrystalline spinels as oxygen reduction and evolution electrocatalysts[J]. Nature chemistry,2011,3(1):79-84.
[21] RIOS E, GAUTIER J L, POILLERAT G, et al. Mixed valency spinel oxides of transition metals and electrocatalysis: case of the MnxCo3-xO4system[J]. Electrochimica acta,1998,44(8/9):1491-1497.
[22] AN L, XIA Z, CHEN P, et al. Layered transition metal oxynitride Co3Mo2OxN6-x/C catalyst for oxygen reduction reaction[J]. ACS applied materials & interfaces,2016,8(43):29536-29542.
[23] LIN H, LIU N, SHI Z, et al. Cobalt-doping in molybdenum-carbide nanowires toward efficient electrocatalytic hydrogen evolution[J]. Advanced functional materials,2016,26(31):5590-5598.
[24] LU Y, JIANG Y, GAO X, et al. Strongly coupled Pd nanotetrahedron/tungsten oxide nanosheet hybrids with enhanced catalytic activity and stability as oxygen reduction electrocatalysts[J]. Journal of the American chemical society,2014,136(33):11687-11697.
[25] TONG X, XIA X, GUO C, et al. Efficient oxygen reduction reaction using mesoporous Ni-doped Co3O4nanowire array electrocatalysts[J]. Journal of materials chemistry A,2015,3(36):18372-18379.
[26] MAYRHOFER K J J, STRMCNIK D, BLIZANAC B B, et al. Measurement of oxygen reduction activities via the rotating disc electrode method: from Pt model surfaces to carbon-supported high surface area catalysts[J]. Electrochimica acta,2008,53(7):3181-3188.
[27] ZHU L D, ZHAO T S, XU J B, et al. Preparation and characterization of carbon-supported sub-monolayer palladium decorated gold nanoparticles for the electro-oxidation of ethanol in alkaline media[J]. Journal of power sources,2009,187(1):80-84.
[28] SEO M H, CHOI S M, KIM H J, et al. The graphene-supported Pd and Pt catalysts for highly active oxygen reduction reaction in an alkaline condition[J]. Electrochemistry communications,2011,13(2):182-185.
[29] STAYKOV A, T LLEZ H, AKBAY T, et al. Oxygen activation and dissociation on transition metal free perovskite surfaces[J]. Chemistry of materials,2015,27(24):8273-8281.
[30] HUANG Y J, DAI H H, LI W S, et al. Electrodeposition preparation of Pt-HxWO3composite and its catalytic activity toward oxygen reduction reaction[J]. Journal of power sources,2008,184(2):348-352.

