3-羟基-2-吲哚酮的电化学合成研究
2018-05-08,,,,
,, ,,
(1.浙江工业大学 化学工程学院,浙江 杭州 310014;2.浙江工业大学 绿色化学合成技术国家重点实验室培育基地,浙江 杭州 310014)
吲哚类衍生物常用于制备药物及染料中间体[1-2],具有抗肿瘤[3]、消炎[4]、抗菌[5]、生物激酶抑制剂[6]和生物抗氧化活性[7]等功能.根据原料进行区分,以取代苯环为原料,包括Fisher法[8]、Reissert法[9]、L-B法[10]和Meerwein芳基化法[11];以吲哚[12]或取代吲哚为原料,进行官能团的修饰.3-羟基-2-吲哚酮是重要的药物中间体,常以靛红为原料,其合成方法包括:1) 以雷尼镍为催化剂,在压力1.5 MPa和温度25 ℃下通入高纯氢气,反应15 min可得产率为84.0%[13];2) 以NaBH4为还原剂,在由EtOH和CH2Cl2组成的混合溶剂中,于0 ℃反应可得产率为75.0%[14];3) 以Na2S2O6还原剂,产率为73.0%[15].上述方法中,靛红廉价易得,但添加大量还原剂提高了处理成本,不易控制产物的选择性.
有机电合成是一种安全环保、选择性良好的绿色合成方法[16-17],电子作为清洁的催化剂代替污染性的还原剂,试剂成本低廉,对反应设备要求低,完全适用于3-羟基-2-吲哚酮的合成.采用电化学还原法制备目标产物,以靛红为原料,硫酸和盐酸等无机酸为支持电解质,有机水溶液为反应溶剂,Cu为工作电极,研究了电解液组成对电还原过程的影响.在此基础上,以Cu为工作电极,Pt为辅助电极,进行电解合成,研究了反应温度和电势对反应电解效果的影响,并以Pb为工作电极进行对照试验,这为以靛红制备吲哚衍生物提供了新思路.
1 实验部分
1.1 试剂、原料和仪器设备
试剂:硫酸、盐酸、磷酸;甲醇、乙酸乙酯、石油醚、1,4-二氧六环、无水乙醇、二甲基亚砜(DMSO)和N,N-二甲基甲酰胺(DMF),以上试剂均为分析纯;去离子水(电阻为15 MΩ);铂片(纯度>99.9%),铜片(纯度>99.5%),铅片(纯度>99.9%);Nepem-41(PTFE)全氟磺酸离子交换膜(贝斯特工贸有限公司);靛红(Isatin,分析纯,上海阿拉丁生化科技股份有限公司).
分析测试仪器:电化学工作站,CHI660E,上海辰华仪器公司;全数字化傅立叶超导核磁共振谱仪,Bruker AVANCE Ⅲ 500 MHz,瑞士Bruker公司;旋转蒸发器,RE-2000,杭州瑞佳精密科学有限公司;集热式恒温加热磁力搅拌器(DF-101S型),巩义市予华仪器有限责任公司.
1.2 电合成装置
电解池为三电极可拆卸玻璃槽.阴、阳极室容积相同,两室之间使用Nepem-41(PTFE)全氟磺酸离子交换膜作为隔膜[18].阳极室采用Pt片为辅助电极(有效面积1.0 cm2),阴极室分别以高纯Cu片或Pb片为工作电极(有效面积1.0 cm2),饱和甘汞作为参比电极,装置如图1所示.

图1 电解槽结构示意图Fig.1 Electrolyze structure diagram
1.3 实验过程
1.3.1 靛红的线性扫描伏安测试
以Cu为工作电极,Pt为辅助电极,饱和甘汞为参比电极,分析有机溶剂、无机酸、溶剂含量、酸浓度等对靛红电还原过程的影响.测试前通N2除去溶解氧.阳极电解液均为50.0 mL的1.0 mol/L硫酸水溶液,阴极电解液的配制如下:
1) 有机溶剂.分别配制含20%相同体积分数的甲醇、乙醇、DMF、DMSO和1,4-二氧六环5种阴极液,以及组分仅为硫酸-水溶液的空白组,共6组.
2) 无机酸.配制含5.0 mol/L甲醇,1.0 mol/L H2SO4,0.01 mol/L靛红的阴极液,再分别配制无机酸为HCl和H3PO4的两种阴极液.
3) 甲醇浓度.分别配制含2.5,5.0,7.4,9.9,12.4,14.9 mol/L甲醇的阴极液,在50.0 mL阴极液中依次对应甲醇体积分数为10%,20%,30%,40%,50%,60%.
4) H2SO4浓度.分别配置含1.0,1.1,1.2,1.3,1.4,1.5 mol/L H2SO4的阴极液.
1.3.2 电化学还原反应
3-羟基-2-吲哚酮的电合成反应机理分别为
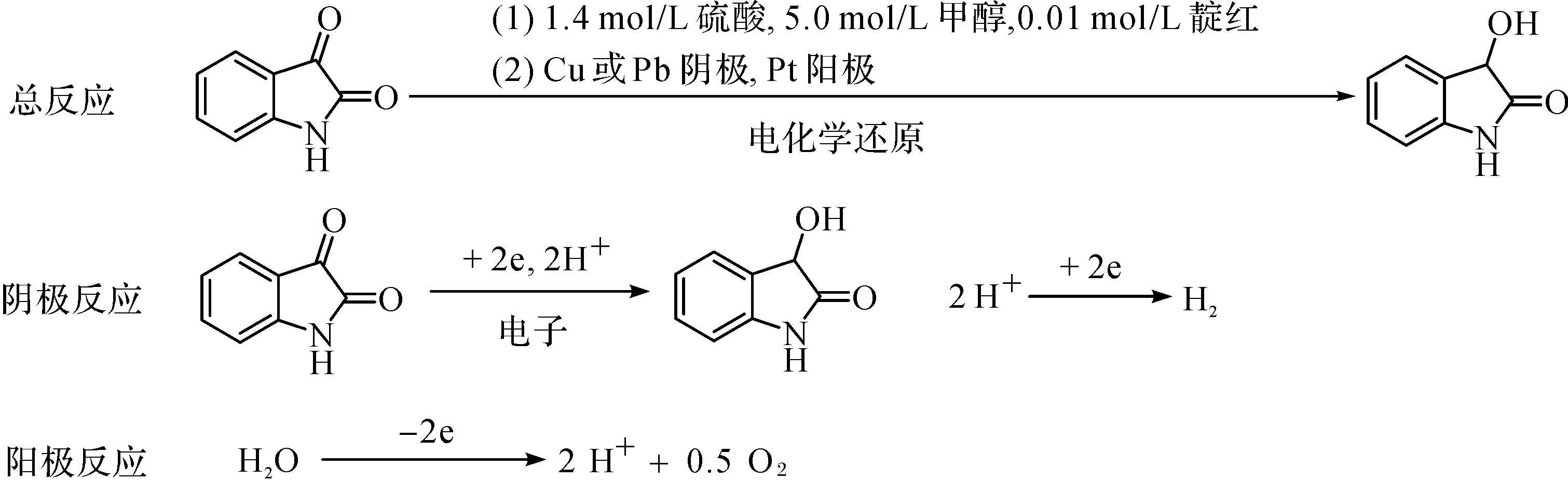
采用恒电位电解法进行电合成.根据线性扫描伏安结果,选择合适的电解液.以Cu为工作电极,Pt为辅助电极,将反应器置于恒温水浴锅中,在不同反应温度、电势和时间下进行恒电位电解;另以Pb为工作电极进行对照实验.反应结束后,往阴极液中添加稀氨水至pH为5~6,用乙酸乙酯多次萃取分离,浓缩有机相,对粗产物进行柱层析分离或重结晶,得淡黄色固体3-羟基-2-吲哚酮.
1.3.3 产物分析
反应过程中通过薄层色谱(TLC)追踪反应状况,反应结束后通过核磁共振波谱仪对产物进行表征.
产物收率的计算可表示为
(1)
电流效率的计算可表示为
(2)
式中:A为产物的摩尔质量(相对分子质量);F为法拉第常数;I为电解电流;m理,m实分别为生成产物的理论质量和实际质量;n为反应电子数;Q理,Q实分别为生成一定量产物所必需的理论电量和实际消耗的总电量;t为电解时间;Y为产物收率;η为电流效率.
靛红(Isatin):mp.202~203 ℃.1H NMR(500 MHz, DMSO-d6) δ 11.04(s,1H),7.59(td,J=7.7,1.4 Hz,1H),7.51(d,J=7.5 Hz,1H),7.07(td,J=7.5,0.9 Hz,1H),6.91(d,J=7.9 Hz,1H).13C NMR(126 MHz, DMSO-d6) δ 184.36,159.33,150.70,138.35,124.65,122.74,117.79,112.19. IR(KBr)ν/cm-1:3 192.5,3 111.3,3 056.5,3 037.2,2 889.1,1 728.3,1 616.6,1 461.2,1 332.0,1 201.6,1 095.5,946.1,770.8,661.5,456.0.
3-羟基-2-吲哚酮(3-hydroxy-2-indolinone):mp.166~167 ℃.1H NMR(500 MHz, DMSO-d6) δ 10.23(s,1H),7.28(d,J=7.3 Hz,1H),7.20(tt,J=7.7,1.1 Hz,1H),6.96(td,J=7.5,1.0 Hz,1H),6.79(d,J=7.8 Hz,1H),6.17(d,J=7.6 Hz,1H),4.83(d,J=7.5 Hz,1H).13C NMR (126 MHz, DMSO-d6) δ 177.93,142.17,129.30,128.91,124.76,121.49,109.47,69.17. IR(KBr)ν/cm-1:3 420.7,3 184.3,3 059.9,2 921.3,2 853.7,1 703.6,1 624.3,1 469.6,1 353.7,1 269.7,1 200.9,1 109.2,1 073.8,749.3,656.3,496.6.
2 实验结果与讨论
2.1 线性扫描伏安结果
以Cu为工作电极,Pt为辅助电极,饱和甘汞为参比电极,在30 ℃下探讨了有机溶剂、无机酸、溶剂含量和酸浓度等反应参数对靛红电还原过程的影响.
2.1.1 有机溶剂对靛红电还原的影响
从图2中可知:对比空白组,在-0.10~-0.50 V,存在电化学窗口较宽的靛红还原峰.甲醇、乙醇和DMF对应着相近的伏安曲线,1,4-二氧六环和DMSO因峰电流密度低,故不合适.当电势小于-0.50 V时,甲醇和乙醇体系析氢较强,适当强度的析氢有利于营造还原氛围,但是DMF易燃、毒性大,甲醇的介电常数又高于乙醇.综合考虑,选甲醇作为有机溶剂.

图2 不同溶剂下电势和电流密度的关系Fig.2 Relationship between potential and current density at different organic solvents
2.1.2 无机酸对靛红电还原的影响
从图3中可知:随着电势的负移,相同电势下H2SO4体系对应的电流密度高于H3PO4体系,且对应起始峰电位更正,表明H2SO4体系对靛红的电还原活性高.在HCl体系中,还原峰虽明显,但HCl会和原料成盐,影响靛红3-羰基的电还原.故选择H2SO4作为支持电解质合适.

图3 不同无机酸下电势和电流密度的关系Fig.3 Relationship between potential and current density at different inorganic acid
2.1.3 甲醇浓度对靛红电还原的影响
从图4中可知:在-0.10~-0.50 V区间内,甲醇浓度为5.0~12.4 mol/L时体系对应的峰电流密度较接近,2.5 mol/L和14.9 mol/L甲醇对应的峰电流密度明显偏低.相同电势下,5.0 mol/L体系对应的峰电流密度高于7.4 mol/L和9.9 mol/L体系,低于12.4 mol/L体系.由于5.0 mol/L体系对应的起始峰电位最正,且当电势小于-0.50 V时,5.0 mol/L体系和12.4 mol/L体系析氢接近.考虑到试剂成本和5.0 mol/L甲醇已足够溶解原料,故选择5.0 mol/L甲醇.
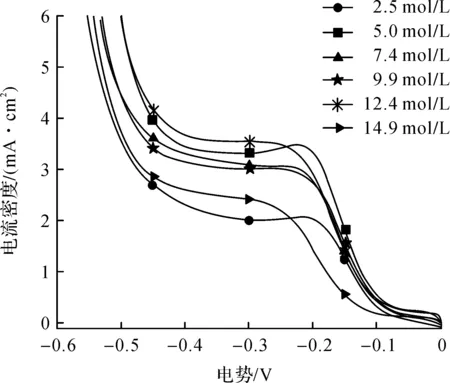
图4 不同甲醇浓度下电势和电流密度的关系Fig.4 Relationship between potential and current density at different concentration of methanol
2.1.4 H2SO4浓度对靛红电还原的影响
从图5中可知:在-0.10~-0.50 V范围内,存在羰基的还原峰,1.0 mol/L和1.5 mol/L体系对应的峰电流密度高于其他体系.当电位小于-0.50 V时,1.0 mol/L和1.5 mol/L体系的峰电流密度较大且相近,而适当的析氢量可以保护靛红结构中的仲胺在反应过程中不被氧化.在相同电势下,虽然1.0 mol/L体系对应的电流密度高于1.5 mol/L 体系,但后者起始峰电位更正,为保证长时间电解合成中有足够的氢离子,仍应选高浓度的H2SO4.故选择1.5 mol/L H2SO4.

图5 不同H2SO4浓度下电势和电流密度的关系Fig.5 Relationship between potential and current density at different concentration of H2SO4
2.2 3-羟基-2-吲哚酮的电化学合成
从上述结果可知:以Cu为工作电极,当阴极液由1.5 mol/L的H2SO4,5.0 mol/L的甲醇和0.01 mol/L的靛红组成,阳极液由1.0 mol/L的H2SO4组成时,能体现良好的电还原效果.在此基础上,研究温度、电势、反应时间对电合成反应的影响,并另以Pb为工作电极进行对比实验,结果如表1所示.恒电位电解过程中电流密度随反应时间的变化存在浮动区间.
2.2.1 Cu电极下产率和电流效率的变化
从图6和表1中可知:当电极电势为-0.9 V、电解时间为4 h、温度为40 ℃时,3-羟基-2-吲哚酮的产率最高,为91.1%;当电极电势为-0.3 V、电解时间为8 h、温度为30 ℃时,电流效率最高,为34.8%.对于反应温度,当电势在-0.3~-0.7 V时,相同电势对应的产率随温度的升高逐渐增大;当电势在-0.9~-1.3 V时,40 ℃体系对应的产率均高于相同电势下其他温度对应的体系;当电势在-0.3~-1.3 V时,相同电势下电流效率随温度的升高均逐渐降低.对于电势,相同温度下的电流密度随还原电势的提高均逐渐增大,反应速度加快,但电势决定了靛红的电还原方向,电势值越负,产物的选择性先升后降,所以当电势负于-0.9 V,产率逐渐降低;相同温度下的电流效率,在-0.3 V时均最高,随电势的负移,析氢逐渐加强,过量氢气阻碍了靛红与Cu电极表面的作用,导致整体呈下降趋势.对于反应时间,当电势在-0.3~-0.7 V时,由于较正电势下电流密度低,将时间由4 h延至6 h或8 h未能提高产率,反而降低了电流效率,而且延长时间对产物的选择性存在影响.

图6 Cu电极下产率和电流效率的变化趋势Fig.6 Variation of yield and current efficiency with Cu
2.2.2 Pb电极下产率和电流效率的变化规律
从图7和表1中可知:当电极电势为-1.1 V、电解时间为4 h、温度为50 ℃时,3-羟基-2-吲哚酮的产率最高,为89.5%;当电极电势为-0.7 V、电解时间为4 h、温度为40 ℃时,电流效率最高,为61.4%.对于反应温度,当电势在-0.5~-1.1 V时,相同电势对应的产率随温度的升高逐渐增大;当电势在-0.3和-1.3 V时,分别为40 ℃体系和30 ℃体系对应的产率最高;当电势在-0.3~-0.5 V和-0.9~-1.3 V时,相同电势下电流效率随温度的升高均逐渐降低,但在-0.7 V时,40 ℃体系高于其他体系,整体而言,升温不利于提高电流效率.对于电势,在-0.3~-1.1 V时,随着还原电势的增大,电流密度提高,反应速度加快,相同温度下的产率均逐渐增大,但在-1.3 V时,由于电势较负,电极还原能力强,降低了靛红3-羰基的选择性还原,产率降低;在-0.3~-0.7 V时,相同温度下的电流效率均先下降,后上升,但在-0.7~-1.3 V时,随电势的负移再逐渐降低.对于反应时间,在-0.3~-0.7 V时,将时间由4 h延至6 h或8 h,未能明显提高产率,电流效率下降,而且电流密度整体偏低,反应速率较慢,析氢不明显.

图7 Pb电极下产率和电流效率的变化趋势Fig.7 Variation of yield and current efficiency with Pb

温度/℃电势/V时间/hCu阴极电流密度/(mA·cm-2)产率/%电流效率/%Pb阴极电流密度/(mA·cm-2)产率/%电流效率/%30-0.383~752.034.81~326.043.6-0.5610~1463.423.63~530.433.9-0.7415~2075.428.94~747.658.0-0.9450~6079.09.66~1165.051.2-1.14110~12572.14.112~1569.534.5-1.34180~20074.52.617~2064.024.240-0.387~1058.022.92~528.527.3-0.5616~2067.616.85~732.824.4-0.7435~4085.415.34~855.061.4-0.9460~7091.19.412~1572.035.7-1.14130~14580.43.916~1878.530.9-1.34190~20075.02.617~2461.320.050-0.385~1462.422.04~627.618.5-0.5627~3572.910.56~935.521.1-0.7450~6087.610.77~1158.243.3-0.9480~9590.46.915~1886.435.1-1.14160~18076.03.021~2589.526.1-1.34220~24070.02.126~3060.114.4
2.2.3 Cu和Pb电解效果的比较
在Cu电极和Pb电极体系中,最高产率分别为91.1%和89.5%,产率相近,但两种电极所得最高产率未分别对应最高的电流效率,说明在提高产物选择性的同时,电势和温度的改变会降低反应的电流效率.对于与最高产率相对应的电流效率,由于Pb电导率低于Cu,导致电流密度低,而且Pb电极析氢过电位较高,氢气析出量较少,电流利用率高,所以电流效率更高.对于温度,Pb电极比Cu电极体系高10 ℃,但反应速率更低,虽然两种电极体系均存在产率随温度的升高而增大的情况,可在高还原电势下,电极电还原活性强,高温反而降低了产物的选择性.对于还原电势,相同反应时间Cu电极体系较Pb正移了0.2 V,说明Cu电极还原活性更强,能耗更低.由于Pb属于重金属,电还原过程中会有少量Pb溶于电解液中,提高了处理成本,结合上述分析,以Cu为工作电极、电极电势-0.9 V(vs. SCE)、电解时间为4 h和温度为40 ℃时,反应条件适宜.
3 结 论
以靛红为原料,采用电化学还原法成功制备了3-羟基-2-吲哚酮,为进一步研究靛红电还原提供了理论和实验依据.Cu和Pb两种电极材料均可用于靛红3-位羰基的电还原,最高产率相近,但Pb电极体系对应更高的温度和还原电势,能耗大,而且Pb电导率低,具有污染性,故工作电极选用Cu适宜.温度和电势对反应影响较大,在合适的还原电势区间内升温才可提高产物的选择性和生成速率.相较传统化学法,电还原中以甲醇-水为溶剂,H2SO4为支持电解质,成本低,污染小,避免了高压下通氢气带来的安全问题和使用大量还原剂造成的污染,符合绿色合成宗旨.
参考文献:
[1] 张江华,吕英,贾红亮,等.吲哚二甲川菁的合成、晶体结构、光谱性质及生物应用[J].高等学校化学学报,2015,36(10):1924-1932.
[2] 刘平.吲哚5位和3位取代化合物的合成研究[D].长沙:长沙理工大学,2012.
[3] 张屹,孟霞,秦大伟,等.吲哚类抗肿瘤药物的研究进展[J].化工中间体,2011(1):15-18.
[4] 张和智,欧阳贵平,陈洪,等.2-吲哚酮类化合物的研究进展[J].精细化工中间体,2016,45(1):9-15.
[5] BHASKAR G, ARUN Y, BALACHANDRAN C, et al. Synthesis of novel spirooxindole derivatives by one pot multicomponent reaction and their antimicrobial activity[J]. European journal of medicinal chemistry,2012,51:79-91.
[6] 祝华建.STR1表达体系优化,新底物发现与新吲哚生物碱酶化学法制备及杂环化合物的构建[D].杭州:浙江大学,2014.
[7] 霍梦月,韦星船,段彦飞,等.一种吲哚基姜黄素类似物的合成及其抗氧化活性研究[J].广东化工,2014,41(9):14-16.
[8] PITCHA P, NEPOLRAJ A, SATHIYASEELAN M, et al. 4-Dihydroxy-3-(indol-2-yl)-quinoline via a substantial methodology-fisher indole synthesis[J]. Heterocyclic letters,2016,6(1):11-14.
[9] UHLE F C. The synthesis of 5-keto -1,3,4,5-tetrahydrobenz[cd] indole; a synthesis of 4-substituted indoles[J]. Journal of the American chemical society,1949,71(3):761-6.
[10] BATCHO A D, LEIMGRUBER W. Intermediates for indoles: US,3976639 A[P].1976-08-24.
[11] RAUCHER S, KOOLPE G A. Synthesis of substituted indoles via meerwein arylation[J]. Journal of organic chemistry,1983,48(12):2066-2069.
[12] RUSSELL H, HARRIS B, HOOD D, et al. 5-substituted
indoles via sodium indoline-2-sulfonate. A reexamination[J]. Organic preparations and proceduresl,1986,17(6):391-399.
[13] PORCS-MAKKAY M, Volk B, KAPOLLER-DEZSOFI R, et al. New routes to oxindole derivatives[J]. Monatshefte fuer chemie,2004,135(6):697-711.
[14] BERGONZINI G, MELCHIORRE P. Dioxindole in asymmetric catalytic synthesis: routes to enantioenriched 3-substituted 3-hydroxyoxindoles and the preparation of maremycin A[J]. Angewandte chemie, international edition,2012,51(4):971-974.
[15] BERGMAN J, STENSLAND B. Cyclization of cyanoethylated ketones as a route to 6-substituted indole derivatives[J]. Journal of heterocyclic chemistry,2014,51(1):1-10.
[16] 马淳安,周晓波.乙醇钛的绿色电化学合成研究[J].浙江工业大学学报,2009,37(3):259-262.
[17] 马淳安,王晓娟,李国华,等.α-硝基萘在Cu-Hg电极上的电化学行为[J].浙江工业大学学报,2008,36(2):119-121.
[18] 刘小建,苑会林.氟塑料薄膜在光伏电源和燃料电池中的应用-PVDF电池背板膜及全氟磺酸离子交换膜的制作与应用[J].塑料工业,2011,38(S1):109-115.
