ABNO催化的伯胺自氧化偶联合成亚胺
2018-05-08,
,
(浙江工业大学 化学工程学院,浙江 杭州 310014)
由于C=N双键具有亲电性,亚胺能参与到一系列的有机反应中,如环加成、环氧化和缩合反应等[1].亚胺也是一类重要的化合物,其已被广泛应用于生物医药、农药、功能材料和缓解腐蚀等领域[2-4].因此,亚胺类化合物的合成和应用研究一直都是有机化学研究的中心内容之一.醛或酮与胺缩合是合成亚胺类化合物的经典方法,但是该方法经常需要使用路易斯酸、碱和脱水剂等[5-7].在过去十多年中,化学工作者们已经开发多种用于合成亚胺的方法[8],例如醇与胺的氧化缩合反应、伯胺的自氧化偶联反应和仲胺的氧化脱氢反应等.其中,伯胺自氧化偶联合成亚胺的方法由于只需要一种原料,原子经济性高、操作简单和原料廉价易得等原因而得到广泛的研究[9-14].然而在已有的文献报道中,该合成方法也存在诸多问题,如催化剂配体价格昂贵、需要添加强氧化剂和过渡金属催化剂等.因此,在伯胺自氧化偶联合成亚胺的研究中,提出相应的改进方法,以解决所存在的问题,仍是该研究的难点和挑战.
近年来,ABNO作为一种温和的催化剂在氧化反应中的作用越来越受到人们的重视.另外,空气是一种极为丰富的资源,而空气中的氧气是一种非常绿色经济的终端氧化剂[15-17].笔者尝试以ABNO为催化剂,在空气氛围中催化伯胺的自氧化偶联反应合成亚胺.
1 实验部分
1.1 实验试剂及仪器
采用Agilent 7890A气相色谱仪对产物进行分析,色谱柱为DB-5(30 m×0.25 mm×0.33 μm),采用FID检测器.进样口温度为280 ℃,载气为氮气,其流量为1 mL/min.所选用的测试条件为:柱温100 ℃,保持2 min,15 ℃/min程序升温至280 ℃,恒温15 min,分流比为30∶1.气质检测采用Thermo Trace ISQ气质联用仪,质谱检测条件为传输线温度250 ℃,离子源温度250 ℃.EI离子化方式为电子能量70 eV,其质荷比范围40~400.核磁共振仪(500 MHz)为Bruker DRX 500.另外,笔者所使用的试剂若无特别说明,均为市售CP或AR级,不经特别处理,直接使用.ABNO根据文献[18]的方法合成.
1.2 实验方法
ABNO催化的伯胺自氧化偶联合成亚胺反应的典型操作步骤:在35 mL的玻璃封管中,加入0.214 g苄胺(Ⅰa)(2 mmol),0.014 g ABNO(0.1 mmol),甲苯0.5 mL,用橡胶塞密封管口,上接空气气球,100 ℃下反应,利用GC或TLC监测反应.24 h后,反应结束,反应液直接经硅胶柱层析分离,得到纯净产物(Ⅱa).产物结构通过1H-NMR、13C-NMR和GC-MS进行表征,部分产物表征如下:
N-苄亚甲基苄胺(Ⅱa):分离收率86%;黄色液体;1H NMR (500 MHz, CDCl3) δ 8.44 (s,1H),7.85~7.83 (m,2H),7.48~7.45 (m, 3H),7.39 (d,J=4.5 Hz, 4H),7.34~7.29 (m,1H),4.88 (s,2H);13C NMR (125 MHz, CDCl3) δ 161.95,139.29,136.15,130.74,128.58,128.47,128.26,127.96,126.96,65.03; MS (EI),m/z195.09[M+,10%],91.04 (100%).
N-(4-氟苄亚甲基)-4-氟苄胺(Ⅱb):以4-氟苄胺为原料,合成步骤同Ⅱa.分离收率80%;黄色液体;1H NMR (500 MHz, CDCl3) δ 8.37 (s,1H),7.81~7.79 (m,2H),7.34~7.31(m,2H),7.14~7.11 (m,2H),7.08~7.04 (m,2H),4.79 (s,2H);13C NMR (125 MHz, CDCl3) δ 164.37 (d,J=250.9 Hz), 161.96 (d,J=244.8 Hz), 160.48, 134.96 (d,J=3.1 Hz), 132.34 (d,J=2.8 Hz), 130.14 (d,J=8.7 Hz), 129.44 (d,J=8.0 Hz), 115.69 (d,J=21.9 Hz), 115.27 (d,J=21.3 Hz), 64.11; MS (EI),m/z230.96 [M+, 6%], 109.04 (100%).
N-(4-氯苄亚甲基)-4-氯苄胺(Ⅱc):以4-氯苄胺为原料,合成步骤同Ⅱa.分离收率84%;黄色固体;熔点62~64 ℃;1H NMR (500 MHz, CDCl3) δ 8.36 (s, 1H),7.73 (d,J=8.5 Hz, 2H),7.41 (d,J=8.5 Hz,2H),7.34 (d,J=8.5 Hz,2H),7.28 (d,J=7.8 Hz,2H),4.79 (s,2H);13C NMR (125 MHz, CDCl3) δ 160.84,137.61,136.89,134.47,132.84,129.46,129.27,128.94,128.65,64.18; MS (EI),m/z263.09[M+,8%],125.05 (100%).
N-(4-(三氟甲基)苄亚甲基)-4-(三氟甲基)苄胺(Ⅱd):以4-(三氟甲基)苄胺为原料,合成步骤同Ⅱa.分离收率90%;黄色液体;1H NMR (500 MHz, CDCl3) δ 8.49 (s, 1H), 7.93 (d,J=8.1 Hz, 2H),7.71 (d,J=8.2 Hz,2H),7.64 (d,J=8.1 Hz, 2H),7.50 (d,J=8.0 Hz,2H),4.92 (s,2H);13C NMR (125 MHz, CDCl3) δ 161.11,143.04,139.02,132.60 (q,J=32.6 Hz),129.46 (q,J=32.5 Hz),128.54,128.14,125.67 (q,J=3.8 Hz),125.50 (q,J=3.7 Hz),124.63 (q,J=257.91 Hz),123.46 (d,J=270.41 Hz),64.42; MS (EI),m/z331.09[M+,16%],159.02 (100%).
N-(3-甲基苄亚甲基)-3-甲基苄胺(Ⅱe):以3-甲基苄胺为原料,合成步骤同Ⅱa.分离收率87%;黄色液体;1H NMR (500 MHz, CDCl3) δ 8.41 (s,1H),7.71 (s,1H),7.60 (s,1H),7.37~7.35 (m,1H),7.30~7.28 (m,2H),7.20 (s,2H),7.13 (s,1H),4.84 (s,2H),2.44 (s,2H),2.41 (s,2H);13C NMR (125 MHz, CDCl3) δ 162.06,139.16,138.29,138.07,136.12,131.53,129.15,128.74,128.44,128.37,127.70,125.85,125.06,65.12,21.40,21.23; MS (EI),m/z223.05[M+, 11%],105.07 (100%).
N-(4-甲氧基苄亚甲基)-4-甲氧基苄胺(Ⅱf):以4-甲氧基苄胺为原料,合成步骤同Ⅱa.分离收率86%;黄色液体;1H NMR (500 MHz, CDCl3) δ 8.32 (s,1H),7.74 (d,J=8.8 Hz,2H),7.27 (d,J=8.7 Hz,2H),6.94 (d,J=8.8 Hz,2H),6.90 (d,J=8.7 Hz,2H),4.75 (s,2H),3.86 (s,3H),3.82 (s,3H);13C NMR (125 MHz, CDCl3) δ 161.64,160.89,158.62,131.66,129.78,129.18,129.14,113.94,113.88,64.39,55.33,55.27; MS (EI),m/z255.12[M+,6%],121.04 (100%).
N-(2,4-二甲氧基苄亚甲基)-2,4-二甲氧基苄胺(Ⅱg):以2,4-二甲氧基苄胺为原料,合成步骤同Ⅱa.分离收率81%;黄色液体;1H NMR (500 MHz, DMSO-d6) δ 8.60 (s,1H),7.78 (d,J=8.6 Hz,1H),7.10 (d,J=8.3 Hz,1H),6.61 (d,J=2.3 Hz,1H),6.56 (dd,J=8.8 2.2 Hz, 2H),6.49 (dd,J=8.3,2.4 Hz,1H),4.58 (s,2H),3.84 (s,3H),3.81 (s,3H),3.78 (s,3H),3.75 (s,3H);13C NMR (125 MHz, DMSO-d6) δ 163.18,160.22,160.09,158.25,156.37,130.21,128.27,120.45,117.66,106.53,104.96,98.75,98.52,58.87,56.17,55.85,55.62; MS (EI),m/z315.30[M+, 2%],151.00 (100%).
N-(2-噻吩亚甲基)-2-噻吩甲胺(Ⅱh):以2-噻吩甲胺为原料,合成步骤同Ⅱa.分离收率91%;黄色液体;1H NMR (500 MHz, CDCl3) δ 8.44 (s,1H),7.43 (d,J=5.0 Hz,1H),7.35 (dd,J=3.6,1.0 Hz,1H),7.26 (dd,J=5.0,3.6 Hz,1H),7.10~7.09 (m, 1H),7.03~7.00 (m,2H),4.97 (s,2H);13C NMR (125 MHz, CDCl3) δ 155.36,142.13,141.55,130.92,129.29,127.35,126.85,125.24,124.79,58.48; MS (EI),m/z206.97[M+,7%],97.03 (100%).
N-(3-吡啶亚甲基)-3-吡啶甲胺(Ⅱi):以3-吡啶甲胺为原料,合成步骤同Ⅱa.分离收率84%;黄色液体;1H NMR (500 MHz, CDCl3) δ 8.87 (s,1H),8.62 (dd,J=4.8,1.7 Hz,1H),8.57 (d,J=1.8 Hz,1H),8.49 (dd,J=4.8,1.5 Hz,1H),8.43 (s,1H),8.12 (dt,J=7.9,1.9 Hz,1H),7.66~7.62 (m,1H),7.34~7.31 (m,1H),7.28~7.22 (m,1H),4.81 (s,2H);13C NMR (125 MHz, CDCl3) δ 159.66,151.67,150.20,149.18,148.45,135.49,134.55,134.30,131.28,123.62,123.39,62.34; MS (EI),m/z197.00[M+,3%],64.94 (100%).
N-(1-苯亚乙基)-1-苯基乙胺(Ⅱj):以1-苯乙胺为原料,合成步骤同Ⅱa.分离收率62%;黄色液体;1H NMR (500 MHz, DMSO-d6) δ 7.89~7.87 (s,2H),7.48 (d,J=7.3 Hz, 2H),7.42~7.40 (m,3H),7.35~7.32 (m, 2H),7.24~7.20 (m,1H),4.9~4.87 (m,1H),2.27 (d,J=0.6 Hz,3H),1.42 (d,J=6.5 Hz,3H);13C NMR (125 MHz, DMSO-d6) δ 162.47,146.21,140.71,129.43,128.21,128.04,126.59,126.54,126.37,58.90,24.86,14.98; MS (EI),m/z223.17[M+,6%],104.96 (100%).
2 结果与讨论
2.1 反应条件优化
为了确定最佳反应条件,以苄胺作为模型底物,分别考察了溶剂、温度和ABNO用量对反应的影响,产物的收率通过气相测定.
2.1.1 溶剂对反应的影响
在偶联反应中,溶剂对反应体系具有重要的影响,在不同溶剂中催化剂会表现出不同的反应活性,有的甚至决定反应能否进行.首先对反应的溶剂进行了一系列的筛选,当苄胺投料量为2 mmol,ABNO用量为0.10 mmol,反应温度为100 ℃,空气氛围中反应时间为24 h时,反应结果见表1.

表1 不同溶剂对反应的影响Table 1 The effect of different solvents on the reaction
从表1可以看出:溶剂对反应的影响显著.当溶剂为甲苯和氯苯时,产物亚胺的收率分别为95%和90%(序号1和2);当溶剂为1,4-二氧六环时,产物亚胺收率仅有30%(序号3);而溶剂为N,N-二甲基甲酰胺时,产物亚胺收率为64%(序号4);当溶剂为二甲基亚砜时,产物亚胺收率只有40%(序号5).通过上述实验对比,最终选择甲苯作为溶剂,并在此基础上对其他反应条件进行优化.
2.1.2 温度和ABNO用量对反应的影响
对伯胺自氧化偶联合成亚胺类化合物来讲,需要选择一个合适的反应温度.因此对不同的温度进行了考察,同时,笔者试图减少ABNO用量,研究是否能达到相同的实验结果,其结果见表2(溶剂为甲苯).
表2温度和ABNO用量对反应的影响
Table2TheeffectofdifferenttemperatureandtheamountofABNOonthereaction

序号ABNO/mmol反应温度/℃反应时间/h产率/%10.10100249520.1090248830.1080245440.0810024885—10024trace
从表2可以看出:当反应温度从100 ℃降到90 ℃时,产物亚胺的收率有所降低为88%(序号2);而温度降至80 ℃时,产物亚胺的收率只有54%(序号3);同样当ABNO用量降到0.08 mmol时,产物亚胺的收率为88%(序号4);同时也做了空白实验,不加ABNO时,只检测到微量产物(序号5).因此,最终确定ABNO的用量为0.10 mmol.
经过上述实验的对比分析,最终确定最佳反应条件为:苄胺(2 mmol),ABNO(0.10 mmol),甲苯为溶剂,100 ℃下空气氛围中反应24 h.
2.2 底物拓展
在上述最优反应条件下,笔者对反应的普适性进行了考察,反应结果如表3所示.伯胺自氧化偶联反应式为

表3 不同亚胺的合成1)Table 3 The synthesis of different imines
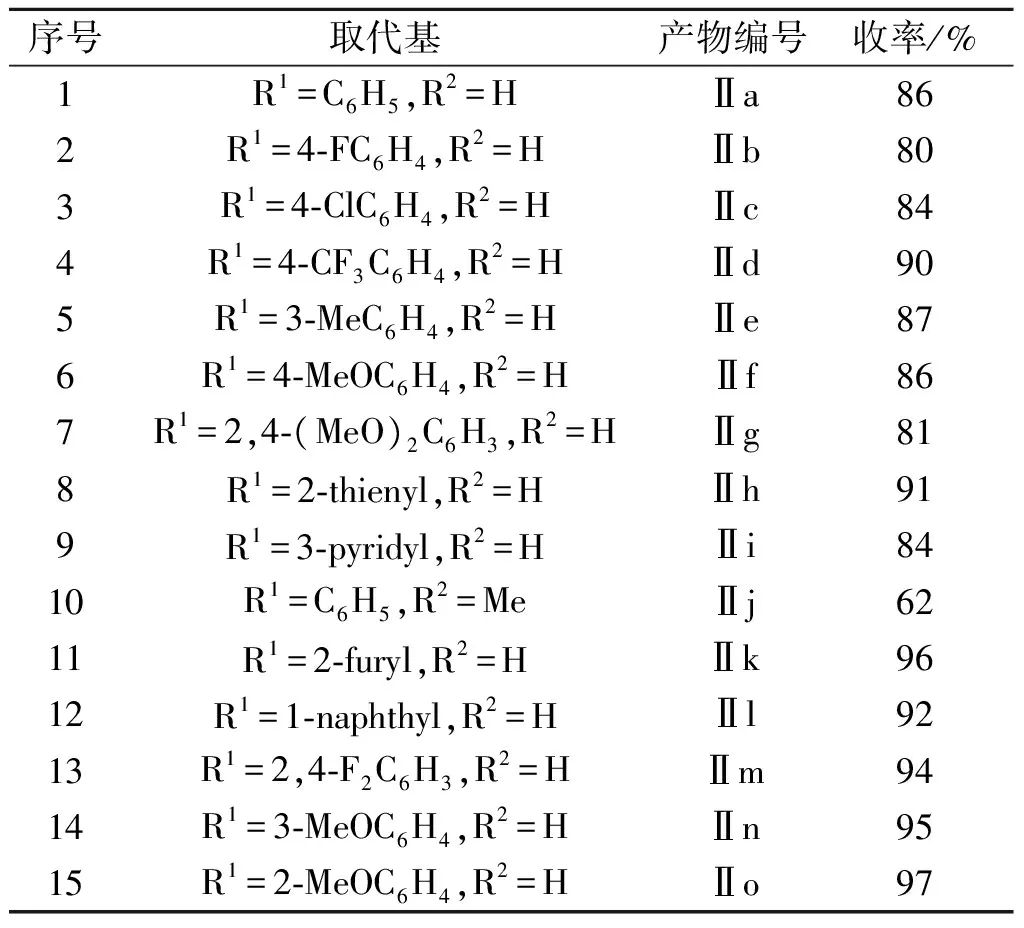
序号取代基产物编号收率/%1R1=C6H5,R2=HⅡa862R1=4⁃FC6H4,R2=HⅡb803R1=4⁃ClC6H4,R2=HⅡc844R1=4⁃CF3C6H4,R2=HⅡd905R1=3⁃MeC6H4,R2=HⅡe876R1=4⁃MeOC6H4,R2=HⅡf867R1=2,4⁃(MeO)2C6H3,R2=HⅡg818R1=2⁃thienyl,R2=HⅡh919R1=3⁃pyridyl,R2=HⅡi8410R1=C6H5,R2=MeⅡj6211R1=2⁃furyl,R2=HⅡk9612R1=1⁃naphthyl,R2=HⅡl9213R1=2,4⁃F2C6H3,R2=HⅡm9414R1=3⁃MeOC6H4,R2=HⅡn9515R1=2⁃MeOC6H4,R2=HⅡo97
注:1) 反应条件:2 mmol底物,0.10 mmol ABNO,0.5 mL甲苯在100 ℃下套空气气球反应24 h,收率为分离收率;序号11~15为气相收率.
从表3可以看出:苄胺自氧化偶联得到亚胺产物气相收率为95%,而分离收率为86%(序号1),可能是由于亚胺不稳定,在过柱纯化过程中易分解,导致分离收率偏低.对于具有不同取代基的苄胺,取代基的电子效应对反应基本没有太大影响.吸电子基取代的苄胺如4-氟苄胺、4-氯苄胺和4-三氟甲基苄胺在0.10 mmol ABNO催化下,相应的亚胺产物分离收率为80%~90%(序号2~4).同样,供电子基取代的苄胺如3-甲基苄胺、4-甲氧基苄胺和2,4-二甲氧基苄胺在0.10 mmol ABNO催化下,相应的亚胺产物分离收率为81%~87%(序号5~7).此外,将该体系应用于杂芳香苄胺如2-噻吩甲胺和3-吡啶甲胺时,反应非常顺利,分别得到了91%和84%的分离收率(序号8,9).当底物是1-苯乙胺时,相应的亚胺产物分离收率为62%(序号10),可能是由于位阻效应的影响使得反应变得困难.此外,其他底物例如糠胺、1-萘甲胺、2,4-二氟苄胺、3-甲氧基苄胺和2-甲氧基苄胺在该体系下也能顺利反应,产物气相收率大于90%(序号11~15),但是这些产物在分离提纯过程中极易分解成醛和胺,难以得到纯品.我们进一步尝试了以两个不同的苄胺类化合物为原料合成不对称亚胺,但产物的选择性并不好.
2.3 实验机理探索
为了探究可能的反应机理,笔者利用连二亚硫酸钠还原了ABNO,制备了ABNOH.ABNOH在空气氛围中100 ℃下反应2 h后,反应液从无色变成红色,此现象表明ABNOH转化成了ABNO,随后柱层析分离得到收率为36%的ABNO.此外,直接以0.1 mmol ABNOH催化2 mmol苄胺的自氧化偶联,得到气相收率为90%的N-苄亚甲基苄胺.结合文献[19-20]和上述实验,我们推测了可能的反应机理为
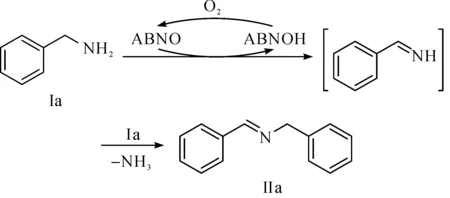
首先苄胺被ABNO氧化成中间体苯甲亚胺,该中间体再与苄胺缩合得到亚胺产物.苄基上的H转移到ABNO上,ABNO转化为ABNOH.ABNOH又被空气中的氧气氧化为ABNO,从而实现整个催化体系的循环.
3 结 论
以ABNO为催化剂,空气为氧化剂,伯胺发生自氧化偶联反应生成亚胺,与其他方法相比,该催化氧化的方法具有简单、高效和选择性高等优点.通过对反应条件的优化,探索出最佳的反应条件:苄胺投料量为2 mmol,催化剂ABNO用量为0.10 mmol,甲苯为溶剂,100 ℃下空气气氛中反应24 h.在优化的条件下,吸电子基或供电子基取代的苄胺、杂芳香苄胺均能顺利反应并得到相应的亚胺产物,分离收率为62%~91%.最后,结合参考文献和相关实验,推测出反应的可能机理.
参考文献:
[1] ADAMS J P. Imines, enamines and oximes[J]. Journal of the chemical society perkin transactions 1,2000,2(2):125-139.
[2] 时蕾,刘艳春,王振平,等.8-羟基喹啉-7-醛缩酰肼席夫碱的绿色合成及抗肿瘤活性研究[J].化学通报,2013,76(1):72-76.
[3] 张欣,覃章兰,肖蒙.含三氮唑环和噻吩环希夫碱的合成及其杀菌活性[J].农药学学报,2005,7(4):353-356.
[4] 闫莹,李伟华,张杰,等.一种席夫碱杂环类碳酸钢酸洗缓蚀剂及其应用:101191226A[P].2008-06-04.
[5] BILLMAN J H, TAI K M. Reduction of schiff bases. II. benzhydrylamines and structurally related compounds[J]. Journal of organic chemistry,1958,23:535-539.
[6] TEXIER-BOULLET F. A simple, convenient and mild synthesis of imines on alumina surface without solvent[J]. Synthesis,1985,16(47):679-681.
[7] WESTHEIMER F H, TAGUCHI K. Catalysis by molecular sieves in the preparation of ketimines and enamines[J]. Journal of organic chemistry,1971,36(11):1570-1572.
[8] CHEN B, WANG L, GAO S. Recent advances in aerobic oxidation of alcohols and amines to imines[J]. ACS catalysis,2015,5(10):5851-5876.
[9] ORITO K, HATAKEYAMA T, TAKEO M, et al. Dimerization of anilines and benzylamines with mercury(ii) oxide-iodine reagent[J]. Tetrahedron,1998,29(44):8403-8410.
[10] YUAN Q L, ZHOU X T, JI H B. Efficient oxidative coupling of amines to imines catalyzed by manganese(III) meso-tetraphenylporphyrin chloride under ambient conditions[J]. Catalysis communications,2010,12(3):202-206.
[11] PRADES A, PERIS E, ALBRECHT M. Oxidations and oxidative couplings catalyzed by triazolylidene ruthenium complexes[J]. Organometallics,2011,30(5):1162-1167.
[12] CAMPBELL A N, STAHL S S. Chemoselective organocatalytic aerobic oxidation of primary amines to secondary imines[J]. Organic letters,2012,14(11):2850-2853.
[13] QIU X, LEN C, LUQUE R, et al. Solventless oxidative coupling of amines to imines by using transition-metal-free metal-organic frameworks[J]. Chemsuschem,2014,7(6):1684-1448.
[14] GE D, QU G, LI X, et al. Novel transition bimetal-organic frameworks: recyclable catalyst for the oxidative coupling of primary amines to imines at mild conditions[J]. New journal of chemistry,2016,40(6):5531-5536.
[15] 蒋栋,金红卫,杨振平,等.一种合成3H-1,2-苯并二磺酸-3-硫酮的简便新方法[J].浙江工业大学学报,2012,40(4):369-373.
[16] 徐伟,张蔓,李景华.水相中氧气氧化芳香醇制备羰基化合物的研究[J].浙江工业大学学报,2013,41(1):65-67.
[17] 陈书斌,孙健,李景华.铜催化苄醇类化合物的氨氧化反应[J].浙江工业大学学报,2015,43(5):578-581.
[18] SHIBUYA M, TOMIZAWA M, SASANO Y, et al. An expeditious entry to 9-azabicyclo[3.3.1]nonaneN-oxyl (ABNO): another highly active organocatalyst for oxidation of alcohols[J]. Journal of organic chemistry,2009,74(12):4619-4622.
[19] LIU L, WANG Z, FU X, et al. Azobisisobutyronitrile initiated aerobic oxidative transformation of amines: coupling of primary amines and cyanation of tertiary amines[J]. Organic letters,2012,44(14):5692-5695.
[20] CHEN B, WANG L, DAI W, et al. Metal-free and solvent-free oxidative coupling of amines to imines with mesoporous carbon from macrocyclic compounds[J]. Acs catalysis,2015,5(5):2788-2794.
