基于FP树的蛋白质功能预测算法研究
2018-05-07葛凌霄
葛凌霄
(四川大学计算机学院,成都 610065)
0 引言
蛋白质组学是以蛋白质为研究对象,以研究细胞、组织或生物体蛋白质组成及其变化规律的科学,目的是通过分析生物体内的蛋白质的表达模式和功能模式,以此能够生物体与细胞的整体水平阐释生命现象的本质规律。对细胞内的蛋白质进行功能注释是蛋白质组学的一个重要的研究方向,通过对未注释蛋白质的功能预测,能够在生物技术制药、生物治疗、农作物基因改良等领域发挥重要作用。传统的生物实验进行功能注释的方法费时且成本高,因此研究基于蛋白质相互作用网络内的计算方法是当前生物信息学家所面临的重要问题。
1 方法
1.1 蛋白质相互作用网络
随着研究蛋白质相互作用的高通量实验技术的发展,现在已可以获取到大量的蛋白质相互作用数据,这也让我们可以使用统计学来对这些数据集进行数据挖掘。我们以图的思想,将这些复杂的蛋白质之间相互作用关系数据构建成为一张复杂的网络,称为蛋白质相互作用网络(Protein-Protein Interaction Network,简称PPI网络),接着我们就能够使用图论以及复杂网络等研究方法对其研究。蛋白质相互作用网络的定义为细胞内所有蛋白质中任意对蛋白质之间可能发生的相互作用关系的完整集合。
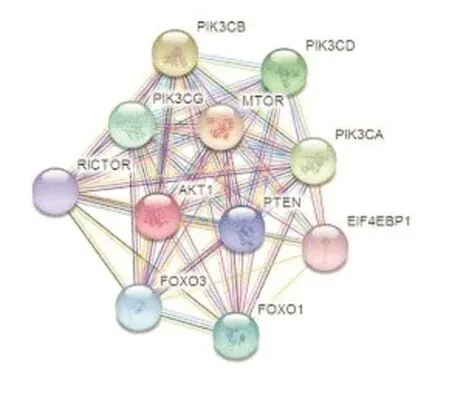
图1
图1是一个蛋白质相互作用网络,我们通常将其看作为一个无向图G(V,E),图中每个节点v表示一个蛋白质,而每条连接两个节点的边e表示蛋白质与蛋白质之间的相互作用关系,在根据具体研究需要时,有时会给边分配权值变为有权图,此时边的权值代表两个蛋白质之间作用关系的强度。
1.2 中心接近度
在现实中,我们经常会遇到求两个点之间最短路径的问题,但也有这样实际生活场景,例如要建造一个大型的娱乐商场,希望光临的顾客到达这个商场的距离都可以尽可能地短。这个就涉及到接近中心性的概念,接近中心性的值为路径长度的倒数。
接近中心性需要考量每个结点到其他结点的最短路径的平均长度。也就是要计算的是到图中其他节点的距离总和比较小,计算的是这个节点处于图中间位置的程度。在一个复杂网络里,接近中心性越高的节点,越趋向于整个图的中心。
在蛋白质相互作用网络里,蛋白质i的接近中心度定义为:
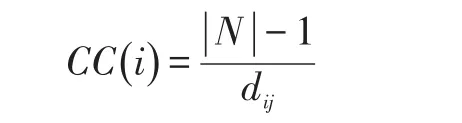
式中,N为蛋白质节点i的邻接节点,为蛋白质节点i与蛋白质节点j的最短路径距离。
1.3 FP树
关联分析通常能够用来挖掘出数据之间的联系,其中最常用的方法就是关联规则的挖掘。FP树,又称FP-growth算法就是关联规则挖掘的常用方法之一。FP-growth算法的思路为,首先压缩数据集,将数据集内所有事务使用FP树这样的数据结构进行表示,接着使用递归的方法将频繁项集依次分解为各自的子问题进行挖掘。
FP-growth算法步骤:
(1)FP树的构建
FP树是一种前缀树,每个节点有三个指针,分别指向父节点,子节点和链接指针。此外,数据结构中还包含有一个头指针表,头指针表中记录每个元素出现的第一个结点,结点中的链接指针将所有相同的元素连接起来。
算法开始时会开始扫描两次数据库,第一次扫描数据库时,列举出所有项,确定1-项频繁集。第二次扫描数据库时,将数据中支持度小于阈值的项删除,然后将这个数据按照刚才项出现次数排序。排序后每个项集都有一个唯一的顺序,这样可以保证后续算法找出所有不重复的频繁项集。然后将这个数据插入到FP树中,并且更新头指针表和链接指针。
(2)挖掘频繁项集
挖掘频繁项集时,从单项集出发每次增加一个元素。对于每一个频繁项集以前缀路径构造一棵FP树,然后向当前的频繁项集中添加一个元素,然后以深度优先的策略递归地进行这个过程直到发现所有频繁项集。
2 数据与实验
2.1 数据
我们用于实验的酵母细胞蛋白质的相互作用数据来自于String数据库(https://string-db.org/cgi/download.pl?UserId=OaGetiAwHwOi&sessionIdGoVW2b711k9A&species_text=Saccharomyces+cerevisiae),共有 6391个蛋白质和2007134条相互作用信息。
功能注释使用的是慕尼黑蛋白质信息中心(MIPS)所制定的功能目录(FunCat)方案,该方案是一种树形层次结构的分类方案,总共包含有28个大类的主要蛋白质功能。酵母的FunCat注释数据源来自于CYGD(Comprehensive Yeast Genome),目前已有功能注释的蛋白质数量为4779个,这些蛋白质包含了17大类的功能注释,我们将没有功能注释的蛋白质从网络中删除,最终得到的酵母细胞蛋白质相互作用网络的节点为4791个,包含406731条相互作用数据。
2.2 实验过程
(1)计算蛋白质相关度阈值
为了提高预测的精度,首先对蛋白质相互作用网络内的蛋白质进行分类,并计算相关性的阈值。
依照接近中心度公式,计算整个网络内每个节点的接近中心度,使用整个网络内所有节点计算中心度的平均值作为筛选阈值,将蛋白质分为高相关度低相关度两类。
(2)修剪子图
使用与待预测蛋白质节点所对应相关度的蛋白质节点组成新的蛋白质相互作用网络,对网络内的每条边计算其边聚数系数,如果边聚数系数小于对应的阈值,则将其删除,最后形成一张新的子图。
(3)挖掘最大频繁项集预测蛋白质功能
在修建过的子图里,找到需要预测的蛋白质节点的所有邻接节点,使用FP-growth算法计算这些蛋白质节点功能的最大频繁项集,求得结果作为预测蛋白质的功能集合。
2.3 结果分析
为了测试和对比我们的是实验结果,我们使用信息检索领域两个常用的评价指标,准确率和召回率,定义如下:
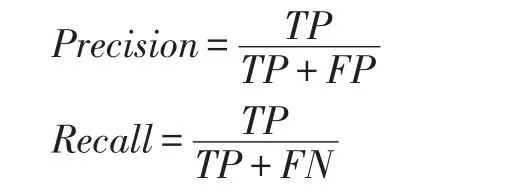
其中TP代表真阳性、FP代表假阳性、FN代表假阴性。
我们将计算出的两个结果与其他两种常用算法进行比较,结果如图2所示,可看出在高相关度下使用FP树进行频繁项集挖掘预测可以提高蛋白质功能预测的准确率。
3 讨论
本文研究的是在蛋白质相互作用网络里对未注释的蛋白质进行功能预测。提出的方法是使用待预测蛋白质在网络中的邻接节点进行关联规则挖掘。在关联分析之前,首先使用复杂网络里接近中心度的思想计算网络内节点的中心性,并计算出阈值,使用阈值对蛋白质节点分类并对网络去边。之后使用FP树来挖掘出邻接节点的最大频繁项集。最终通过实验证明,该算法能够提高蛋白质的功能预测精度。
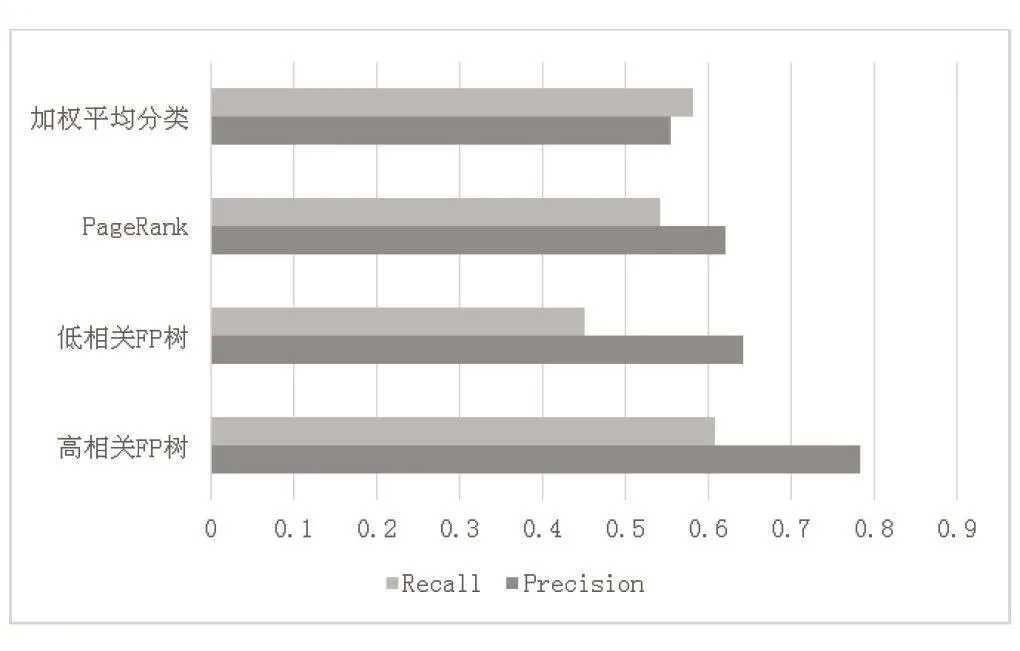
图2
参考文献:
[1]李锦泽,叶晓俊.关联规则挖掘算法研究现状[J].计算机技术与应用进展,2007.
[2]王淑琴.机器学习方法及其在生物信息学领域中的应用[D].吉林:吉林大学,2009
