茶树紫芽中多种花色苷组分同时检测方法的建立
2018-05-03刘林峰
刘林峰
周 阳1,2
林 玲1,2
肖文军1,2,3
龚志华1,2
(1.湖南农业大学茶学教育部重点实验室,湖南 长沙 410128;2.湖南农业大学国家植物功能成分利用工程技术研究中心,湖南 长沙 410128;3.湖南农业大学湖南省植物功能成分利用协同创新中心,湖南 长沙 410128)
花色苷是植物的主要呈色物质[1]。花色苷在茶树中具有普遍性,是茶树抗逆生理的表现形式之一[2]。花色苷具有很好的生物活性[3],成为近年茶叶功能成分研究的热点。茶树芽叶中花色苷分析方法迄今还没有标准,参照其他植物花色苷分析方法[4],其定性分析方法主要有纸层析、薄层层析、紫外-可见吸收光谱[5]等;定量分析方法主要有单一pH值法、差减法、pH示差法及高效液相色谱法等[6-7]。在众多方法中,高效液相色谱法具有高灵敏度、分析速度快等突出优点,且能与其他分析设备联用,如Eva[8]、Chandra[9]等都曾建立高效液相色谱与其他设备谱联用的方法来分析植物中的花色苷。Sakamoto 等[10]也使用HPLC分析哈斯卡普茶花色苷含量。Lee等[11]用pH示差法和HPLC同时分析了含有花色苷的果汁,建立了2种分析方法的相关性。中国高效液相色谱分析植物中花青素也有应用,如张芳轩[12]、江连洲[13]、李琪[14]、徐学玲[15]等分别用HPLC分析了黑米种皮、紫甘薯、酿酒葡萄、紫色小白菜中花色苷组分,说明高效液相色谱是当下较为普遍的分析植物花色苷的方法。
茶叶花色苷的高效分析方法一直在探索中,也有人用HPLC分析茶叶中花色苷组分,龚加顺等[16]采用HPLC分析茶叶中花色苷,初步探究了HPLC分析茶叶花色苷组分。但目前的分析大都是以单一矢车菊为标准品,分析单一的茶树品种。本研究针对上述问题,拟以3种花色苷外标为标准品,建立同时分析多种紫芽茶树花色苷组分的HPLC分析方法,以期为指导紫芽茶树花色苷组分分析、高纯单体的分离鉴定提供基础。
1 材料与方法
1.1 材料
茶样:紫娟、自选9803、红芽佛手、安73,湖南农业大学长安实践教学基地茶树资源圃和湖南省茶叶研究所高桥茶树资源圃,采摘标准为一芽三叶。茶梢冷冻干燥后制成干粉,塑料袋密封后保存于冰箱中,备用。
1.2 试剂及仪器设备
1.2.1 药品试剂
乙腈、磷酸、甲醇、无水乙醇:分析纯,天津市恒兴化学试剂制造有限公司;
乙腈、甲醇:色谱纯,天津市恒兴化学试剂制造有限公司;
标准品矢车菊素-3-O-葡萄糖苷、天竺葵素-3-葡萄糖苷:北京谱析科技有限公司;
飞燕草素-葡萄糖苷:西宝生物科技(上海)股份有限公司;
超纯水:自制;
酸性乙醇水溶液(85 mL无水乙醇加15 mL 1%盐酸水):自配。
1.2.2 仪器设备
旋转蒸发器:SY-2000型,上海亚荣生化仪器厂;
高效液相色谱仪:LC-20A型,日本岛津公司;
茶叶揉捻机:40型,浙江上洋机械股份有限公司;
水浴锅:DSY-2-8型,北京国华医疗器械厂;
冷冻干燥机:MODULYOD-230型,美国Thermo Fisher Sciebtific公司。
1.3 方法
1.3.1 样品花色苷的提取与纯化 参照文献[17~18]和预试验结果,采用回流提取的方法提取紫芽茶树一芽三叶干粉中的花色苷,提取条件为80%乙醇(添加1%冰乙酸水)、1∶50 (g/mL)料液比、80 ℃下回流提取60 min,过滤取滤液,并依次用配比为正己烷∶乙酸乙酯∶水=1∶1∶2(体积比)的溶剂萃取脱脂至有机层无色。水层低温旋转蒸发浓缩,用甲醇定容至2 mL,0.45 μL有机滤膜过膜,装入进样瓶中待HPLC 分析。
1.3.2 标准品溶液的配制 分别取1 mg 3种花色苷标准品,分别用甲醇溶解定容至10 mL;每种标准品溶液各吸取1 mL 混合定容至10 mL,混合均匀,得到混合标准溶液,备用。
1.3.3 茶树紫芽花色苷HPLC分析方法的建立
(1) 检测波长的确定:根据文献[19],用紫外分光光度计对3种标准品溶液进行全波长扫描,选择最适合的分析波长。
(2) 流动相的优化:根据文献[20],控制有机相洗脱梯度为0~55 min、10%~40%,流速 0.8 mL/min,柱温 30 ℃和紫外检测波长 520 nm等其他色谱条件不变,按照表1方案,选用的流动相不同配比,分析比较流动相对样品分离度及柱效的影响。

表1 不同流动相设计方案Table 1 Designs of different mobile phases
(3) 洗脱梯度的调节:根据文献[7],以乙腈和0.2%磷酸超纯水作流动相梯度洗脱,控制流速 0.8 mL/min,柱温 30 ℃,紫外检测波长 520 nm等色谱条件不变,按表2方案,分析不同洗脱梯度下的样品分离度及柱效情况,选择最优的洗脱梯度。

表2 不同洗脱程序设计方案Table 2 Designs of different elution procedures
(4) 流速的选择:根据文献[20],以乙腈和0.2%磷酸超纯水作流动相梯度洗脱,控制有机相洗脱梯度为0~55 min、10%~40%,柱温 30 ℃,紫外检测波长 520 nm等其他色谱条件不变,分析流速(0.8,1.0,1.2 mL/min)对样品分离度及柱效的影响。
(5) 柱温的选择:根据文献[20],以乙腈和0.2%磷酸超纯水作流动相梯度洗脱,控制有机相洗脱梯度为0~55 min、10%~40%,流速 0.8 mL/min,紫外检测波长 520 nm等其他色谱条件不变,分析柱温(20,25,30 ℃)对样品分离度及柱效的影响。
1.3.4 方法学考察
(1) 标准曲线的制作:将1.3.2配制好的混合标准品溶液过膜,用1.3.3中获得的最优色谱条件分别进样5,10,15,20,25,50 μL,绘制标准曲线。
(2) 精密度分析:分别配制3种标准品,分别在最优色谱条件下同日内5个不同时间点进行进样分析,由测得的面积(A)计算标准偏差(SD)和相对标准偏差(RSD),获得日内精密度值。
(3) 重复性分析:按1.3.1的方法,4种紫芽茶树用同一批茶样制备6份不同花色苷提取液在最优色谱条件下进样,由测得的面积(A)计算相对标准偏差(RSD),分析重复性。
(4) 稳定性分析:配制3种标准品的混合标准溶液,室温静置,分别于0,2,4,6,8,12,24 h进样,由测得的面积(A)分析溶液稳定性。
(5) 样品回收率分析:称取 6份已测定了花色苷含量的茶样1 g,按1.3.1 方法制备供试溶液,加入适量混合标准液,在最优色谱条件下进样,重复6次,分析样品回收率。
1.3.5 紫芽茶树茶梢样品花色苷液相色谱法含量分析 按照1.3.1制备4种茶样待测液,进样10 μL,重复3次,计算茶样中花色苷含量。计算公式:
(1)
2 结果与分析
2.1 色谱条件的确定
2.1.1 检测波长的确定 通过HPLC中全波长扫描可知(图1),飞燕草素的紫外最大吸收波长在275,524 nm左右,矢车菊素的紫外最大吸收波长在281,516 nm左右,天竺葵素的最大吸收波长在266,513 nm左右,要同时测定3种标准品以及样品中的花色苷,考虑到各组分的灵敏度并避免其他多酚组分干扰,选择520 nm波长作为检测波长较为合适。

图1 3种标准品紫外可见光谱图Figure 1 UV-visible spectrums of three standard products
2.1.2 流动相的优化 如图2所示,甲醇、50%乙腈、乙腈均可实现各花色苷成分的分离,考虑到乙腈洗脱能力更强,故选择乙腈为有机相。水相磷酸浓度对组分分离影响不大,但在水中加入一定体积的酸,调节流动相的酸碱度可达到减少小峰拖尾和提高分离度的效果,故选择0.2%磷酸水做水相。综上,选择方案4为最佳流动相方案。

图2 不同流动相方案HPLC图Figure 2 HPLC chromatograms of different mobile phase solutions
2.1.3 洗脱梯度的调节 如图3所示,方案1和方案2有机相比率过高,各组分分离度相对较差,方案3耗时过长,与方案4相比效率过低,综合来说方案4花色苷组分分离度和分离效率最佳,故选择方案4,即有机相洗脱梯度为:0~55 min,10%~40%为最佳洗脱方案。
2.1.4 流速的选择 不同流速方案的HPLC图见图4。结果表明,流速越快,虽然会使目标物质的分离时间相对缩短,但样品各组分分离度降低,故选择0.8 mL/min为最适流速。
2.1.5 柱温的选择 不同柱温方案的HPLC图见图5。结果表明,柱温越高,色谱分析出峰时间会更快,但高柱温HPLC调节柱温耗时过多,故选择相对效率较好的30 ℃为系统柱温。
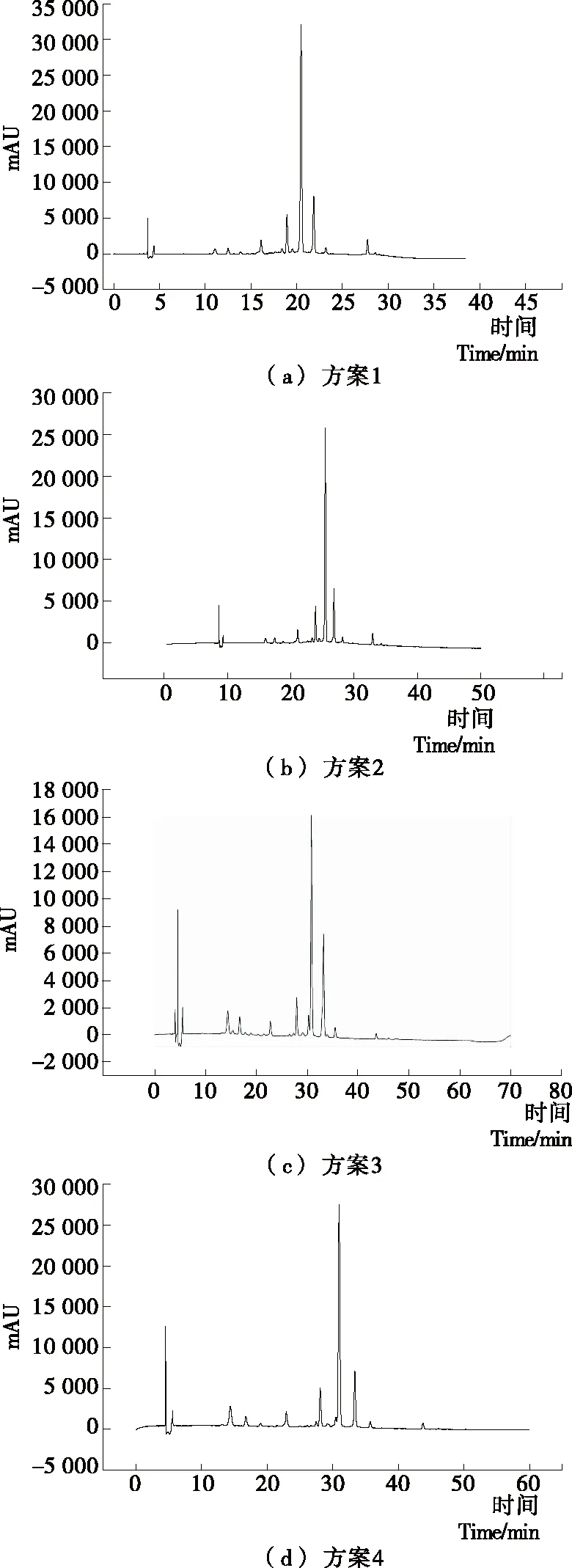
图3 不同洗脱程序方案HPLC图Figure 3 HPLC chromatograms of different elution procedures
2.2 方法学考察
2.2.1 标准曲线的制作与检出限 以进样量为x轴,峰面积为y轴,绘制花色苷混合标准曲线,见表3。由表3可知,在5~75 μg/L范围内,3种标准品都表现出良好的线性关系。
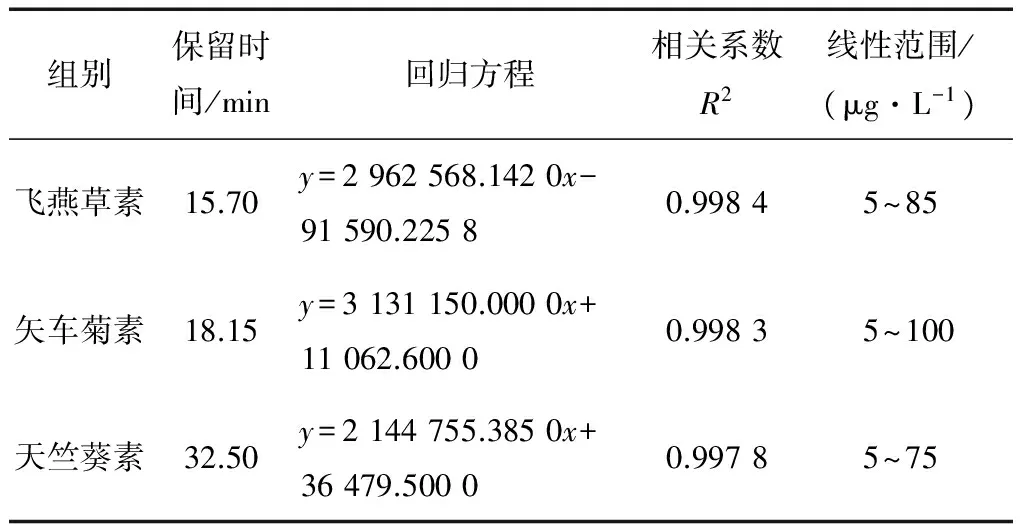
表3 不同花色苷标准品线性关系表Table 3 Linear relationship of different anthocyanin standards

图4 不同流速方案HPLC图Figure 4 HPLC chromatograms of different flow rate
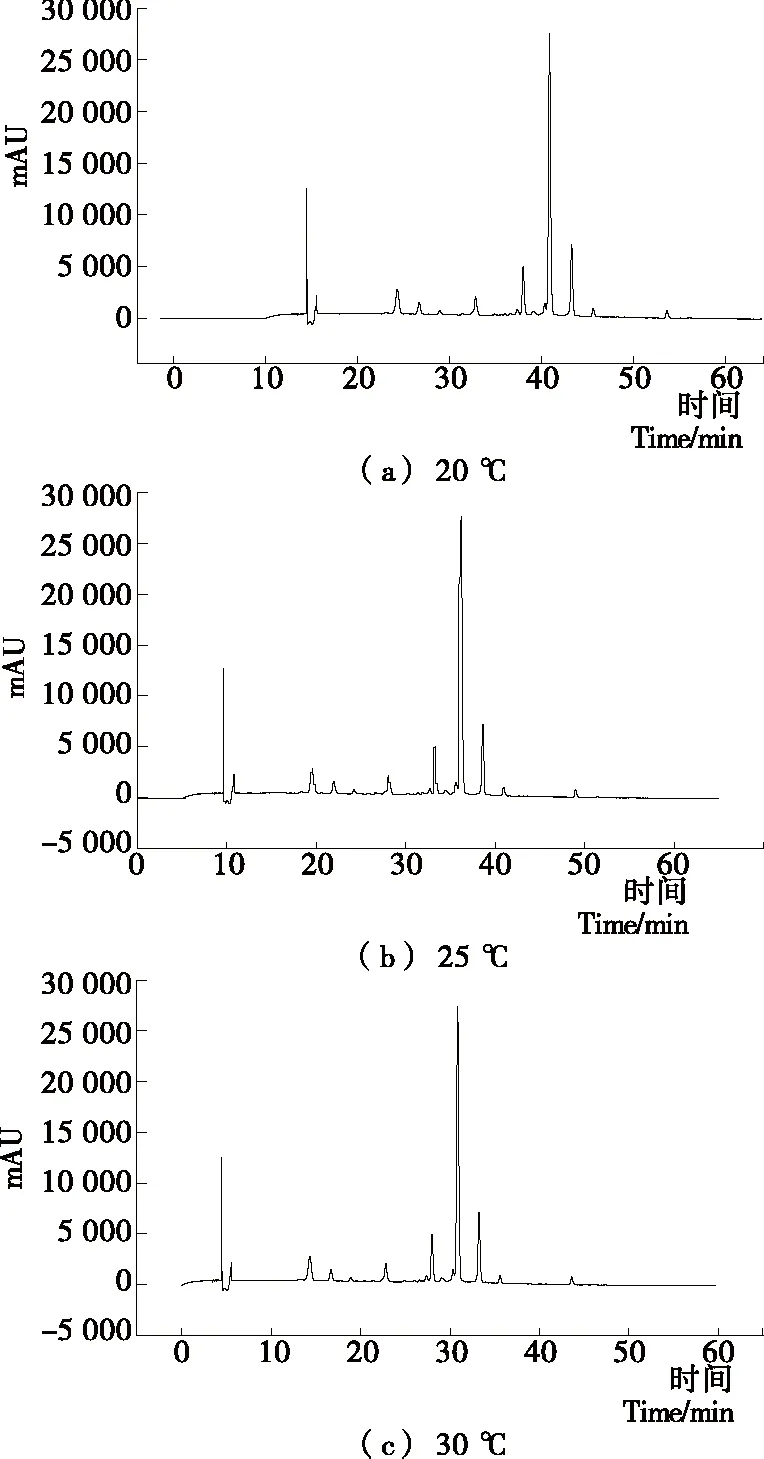
图5 不同柱温方案HPLC图Figure 5 HPLC chromatograms of different column temperature
2.2.2 精密度分析 由表4可知,在520 nm波长下,3种标准品的RSD都在8%内,表明本方法精密度良好。

表4 精密度分析结果Table 4 Results of precision analysis
2.2.3 重复性分析 由表5可知,在520 nm波长下,4种紫芽茶树样品中总花色苷的RSD均在9.04%之内,表明本方法重复性良好。
2.2.4 稳定性分析 由表6可知,室温静置下,520 nm波长下飞燕草素、矢车菊素和天竺葵素的RSD均≤9%,表明本方法稳定性良好。
2.2.5 回收率分析 由表7可知,3种外标花色苷在本方法下加标回收率为75%~88%,到达色谱分析要求,说明本方法结果可靠。
2.3 紫芽茶树茶叶样品中花色苷含量的检测
根据1.3.1中的方法等到的茶树花色苷溶液,按1.3.5的方法重复3次,不同茶样中花色苷含量,结果见表8和图6。由HPLC色谱图可以看出,4种紫芽茶树样品中的7种主要花色苷均得到有效分离。从结果可以看出不同茶树品种(系)所富集的花色苷含量存在显著差异,花色苷含量的高低表现为紫娟>自选9803>安73>红芽佛手,与紫芽茶树品种(系)芽叶发紫程度的深浅基本一致,说明此方法适合分析紫芽茶树中花色苷含量。

表5 重复性分析结果Table 5 Results of repeatability analysis

表6 稳定性分析结果Table 6 Results of stability analysis
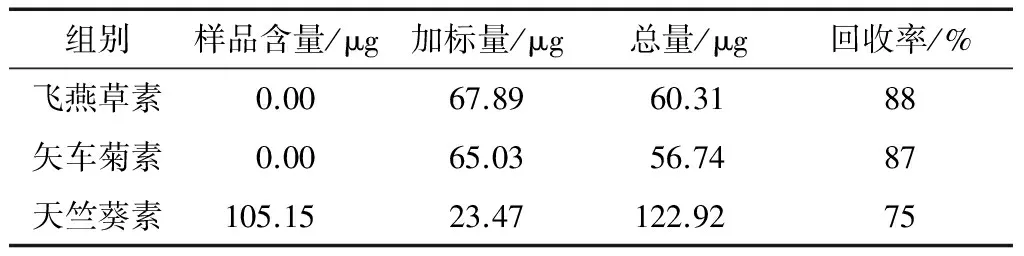
表7 回收率分析结果Table 7 Results of recovery analysis
3 结论
本研究利用高效液相色谱法建立了同时检测4种紫芽茶树花色苷组分的HPLC分析方法:茶梢样品先经酸性80%乙醇水回流提取、正己烷和乙酸乙酯脱脂,然后采用 C18色谱柱,以乙腈和0.2%磷酸超纯水作流动相梯度洗脱,有机相洗脱梯度为0~55 min、10%~40%,流速 0.8 mL/min,柱温 30 ℃,紫外检测波长 520 nm,4种紫芽茶树品种(系)中各花色苷组分和标准品在本方法下分离效果良好,相关系数均≥0.99,线性范围为 5.00~75.00 μg/L,样品加标回收率为75%~88%,该方法精密度、重复性和稳定性的相对标准偏差值≤10%。紫芽茶树检测结果表明,花色苷含量的高低表现为紫娟>自选9803>安73>红芽佛手,与紫芽茶树品种(系)芽叶发紫程度的深浅基本一致。
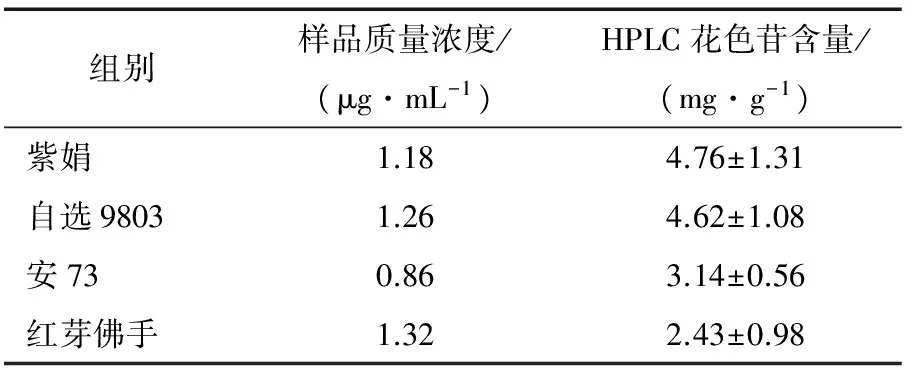
表8 不同样品花色苷含量差异Table 8 Analysis of anthocyaninscontents in different samples (n=3)

图6 不同品种样品和混合标样的花色苷HPLC色谱图Figure 6 Comparison of anthocyanin components differences in different tea varieties and mixed standard
本研究使用的C18为常见色谱柱,购买方便,流动相为乙腈和磷酸水,容易配制,同时也降低了对试验仪器的要求,可用于同时检测分析矢车菊素-3-O-葡萄糖苷、天竺葵素-3-葡萄糖苷、飞燕草素-葡萄糖苷3种植物花色苷和紫芽茶树中多种花色苷组分。
[1] 赵云荣, 王世雷.植物花青素研究进展[J].安徽农业科学, 2008, 36(8): 3 095-3 097.
[2] 杨如兴, 孔祥瑞, 张磊, 等.茶树品种(品系)芽叶色泽表型遗传数量分类研究[J].热带亚热带植物学报, 2016, 24(4): 444-451.
[3] 徐渊金, 杜琪珍.花色苷生物活性的研究进展[J].食品与机械, 2006, 22(6): 154-157.
[4] 孙建霞, 张燕, 孙志健, 等.花色苷的资源分布以及定性定量分析方法研究进展[J].食品科学, 2009, 30(5): 263-268.
[5] LEVI M A, SCARMINIO I S, POPPI R J, et al.Three-way chemometric method study and UV-Vis absorbance for the study of simultaneous degradation of anthocyanins in flowers of the Hibiscus rosa-sinensys species[J].Talanta, 2004, 62(2): 299-305.
[6] 翦祎, 韩舜愈, 张波, 等.单一pH法、pH示差法和差减法快速测定干红葡萄酒中总花色苷含量的比较 [J].食品工业科技, 2012, 33(23): 323-325.
[7] 李子江, 张家国, 刘亚栋.花色苷定性及定量分析方法综述[J].山东商业职业技术学院学报, 2011, 11(6): 99-101.
[8] GARCíABENEYTEZ E, CABELLO F, REVILLA E.Analysis of grape and wine anthocyanins by HPLC-MS[J].Journal of Agricultural & Food Chemistry, 2003, 51(19): 5 622.
[9] CHANDRA A, JATINDER RANA A, LI Yin-qing.Separation, identification, quantification, and method validation of anthocyanins in botanical supplement raw materials by HPLC and HPLC-MS[J].Journal of Agricultural & Food Chemistry, 2001, 49(8): 3 515.
[10] SAKAMOTO M, ARAKAWA Y, MIYOSHI K, et al.Production and Sensory Evaluation of Hokkaido Haskap Tea[J].Nippon Shokuhin Kagaku Kogaku Kaishi, 2012, 59(9): 456-464.
[11] LEE J, RENNAKER C, WROLSTAD R E.Correlation of two anthocyanin quantification methods: HPLC and spectrophotometric methods[J].Food Chemistry, 2008, 110(3): 782-786.
[12] 张芳轩, 张名位, 张瑞芬, 等.黑米种皮中花色苷组成及含量的HPLC分析[J].中国粮油学报, 2010, 25(12): 122-125.
[13] 江连洲, 王晰锐, 张超, 等.HPLC-MS法鉴定不同品种紫甘薯中花色苷组成[J].中国食品学报, 2011, 11(5): 176-181.
[14] 李琪, 李广, 金丽琼, 等.HPLC法测定甘肃地产不同品种酿酒葡萄中的花色苷[J].中国酿造, 2014, 33(3): 132-136.
[15] 徐学玲, 赵岫云, 王丹, 等.HPLC-ESI-MS分析紫色小白菜中花色苷组成分析[J].食品工业科技, 2014, 35(11): 278-281.
[16] 龚加顺, 隋华嵩, 彭春秀, 等.“紫娟”晒青绿茶色素的HPLC-ESI-MS分离鉴定及其稳定性研究[J].茶叶科学, 2012, 32(2): 179-188.
[17] 王秋霜, 凌彩金, 刘淑媚, 等.红紫芽茶叶花青素提取分离及pH稳定性初探[J].中国农学通报, 2014, 30(9): 291-296.
[18] 罗正飞, 梁名志, 王立波.“紫娟”茶花青素提取工艺研究[J].滇西科技师范学院学报, 2011, 33(4): 111-114.
[19] WOLF F T.Absorption spectra of the anthocyanin pigment of red cabbage: a natural wide-range ph indicator[J].Physiologia Plantarum, 2010, 9(4): 559-566.
[20] ARAPITSAS P, SJOBERG P J R, TURNER C.Characterisation of anthocyanins in red cabbage using high resolution liquid chromatography coupled with photodiode array detection and electrospray ionization-linear ion trap mass spectrometry [J].Food Chemistry, 2008, 109(1): 219-226.
