H2O在金红石型IrO2(110)和RuO2(110)表面吸附的DFT分析
2018-04-24周建锋李国荣
周建锋, 李国荣, 王 欣, 唐 电,
(1. 福州大学材料科学与工程学院, 福建 福州 350116; 2. 福州大学材料研究所, 福建 福州 350116)
0 引言
IrO2和RuO2是最重要的电化学催化剂[1], 具有非常优异的电极材料特性, 被广泛应用于电化学工业领域[2-3]. 作为高性能的活性材料, RuO2和IrO2仍有各自的特点. RuO2对析氯反应有很低的过电位, 是优越的析氯催化材料[4], 所以被大量应用于氯碱工业、 氯酸盐工业和次氯酸钠生产. IrO2的催化活性略低于RuO2, 但它具有很高的耐腐蚀性, 尤其在酸性介质中能保持很高的稳定性[5-6], 因此被大量应用于电冶金工业和药剂的生产. RuO2和IrO2不仅在析氯(氧)场合中有大量的应用, 而且由于其优越的储能和能量转换等性能, 因此在水电解、 超电容、 燃料电池和锂电池[7-11]等新能源领域中发挥着作用, 从而使得RuO2和IrO2在近十多年来备受关注. 无论在电化学工业还是在新能源领域应用, RuO2和IrO2的服役环境大多为水溶液, 因此其表面势必会与水发生接触, 吸附的H2O对催化反应进程和效果产生重要影响.Lobo等[12]采用高分辨电子能量损失谱和热脱附仪, 研究认为RuO2的(110)晶面上的H2O分子的吸附,主要于Ru不饱和五配位的吸附位置Rucus(coordinatively unsaturated site)上的吸附, 但由于实验测试方法的限制, 无法获得更深层次的信息, 包括吸附发生后H2O的姿态和方位、 吸附过程以及吸附H2O对 (RuO2)本身结构的影响和吸附体导电性能的变化等. 为弥补实验研究深度的不足, 引入材料计算方法, 加深H2O分子在吸附体表面吸附的机理的认识无疑是非常必要的.
作为催化剂, RuO2和IrO2通常均呈金红石结构, 其(110)晶面是热力学最稳定的晶面. Over等[13]引入材料计算的方法, 不仅证实了RuO2的(110)晶面是最重要的催化晶面, 也揭示该RuO2的(110)晶面在反应中的作用机理. 此后, 有关RuO2(110)和IrO2(110)晶面性质及其催化机理的基础性研究受到重视[14-15]. Kim等[16]证实RuO2的(110)晶面的Rucus位置是活性点, CO和N2分子都倾向于垂直吸附于(110)晶面. Wendt等[17]研究RuO2(110)面对CO的吸附特性和吸附机理. Gong等[18]通过计算, 对比包括Ru、 Ir在内的活性金属及其相应的活性氧化物的催化能力, 认为活性氧化物对CO的氧化的活性更高, 取决于CO-O的键长. Fang等[19]研究RuO2(110)晶面对的析氧反应的催化机制. Hansen等[20]应用第一性原理研究IrO2和RuO2以及其它几种金红石氧化物的(110)晶面的析氯反应机制. Wang等[21-24]应用DFT研究多种基团(包括NHx、 N2、 CH4等)在RuO2(110)和IrO2(110)晶面的吸附原理, 对比氨在RuO2(110)和IrO2(110)晶面氧化的过程. 值得注意的是, 目前尚未查到有关活性氧化物的(110)晶面吸附H2O的计算研究报导. 为深入了解活性氧化物催化剂与吸附水作用的机制, 以及对比IrO2和RuO2这两种重要的催化剂, 本文拟采用第一性原理的计算方法, 尝试对IrO2和RuO2体系的(110)表面吸附H2O的结构和特性进行研究.
1 计算模型与方法
对H2O在IrO2(110)、 RuO2(110)表面吸附进行计算及研究均由VASP软件包完成. 选用基于广义梯度近似(GGA)的交换关联法对能量泛函和单电子K-S方程进行自洽求解, 梯度函数选用PBE. 计算中考虑周期性边界, 价电子波函数采用平面波基组来展开. 截断能量取值为520 eV, 自洽计算过程结束时, 结构的总体能量收敛于5×10-4eV·atom-1, 原子受力标准为每个原子上的力小于0.001 eV·atom-1.
IrO2(110)、 RuO2(110)表面Slab模型, 由优化后的IrO2(110)-(2×2)和RuO2(110)-(2×2)超晶胞而来[15]. 由于IrO2和RuO2都具有相同的金红石型(属P42/mnm点群)结构、 类似的晶格常数及离子半径等, 故以IrO2(110)-(2×2)表面Slab模型为例加以说明. IrO2(110)表面Slab模型由9层原子构成, 如图1(a)所示. 原子堆垛最外层为不饱和2配位桥位Obr(bridge-bonded)原子, 以桥位方式居于6配位Ir6f(six-fold coordinated)原子上方; 第二层以Ir、 O原子混合组成, 包含了6配位Ir6f原子、 不饱和5配位Ircus原子以及3配位平面O3f(three-fold coordinated)原子; 第三层为3配位下桥氧原子. 表面系统真空层厚度取1.5 nm, 经过收敛性测试足以保证Slab模型与周期结构之间无相互作用. Slab模型中表面最外三层原子以及H2O充分弛豫, 其它原子层固定, 直至达到体系的最低能量, 表面原子受力收敛.

图1 IrO2 (RuO2)(110)面Slab和(110)面吸附H2O的模型Fig.1 IrO2 (RuO2)(110) Slab model and H2O absorption on (110)
H2O在IrO2(110)和RuO2(110)表面上的吸附位有Obr、 O3f、 Mecus和Me6f(Me代表Ir或Ru原子,下同)4种. 且H2O吸附时的初始取向, 分别有H2O垂直于表面且氢原子向上(H-up)、 H2O平行于表面 (H-parallel)和H2O垂直于表面且氢原子向下(H-down)3种, 依次如图1 (b)、 1(c)和1(d)所示. 如此H2O在IrO2(110)和RuO2(110)表面共有12种吸附方式.
2 结果与讨论
2.1 结构优化和吸附能
采用1.0 nm×1.0 nm×1.0 nm的原胞优化了自由水分子的结构, 计算得到的键长和键角分别0.097 2 nm和104.52 °, 与文献[25]的水分子结构值0.097 6 nm和105.05°接近.
原子或分子吸附于材料清洁表面时, 吸附后所获得的能量定义为吸附能. H2O吸附于IrO2(110)表面和RuO2(110)表面时, 吸附能计算公式分别定义如下:
(1)

(2)

吸附能可以反映吸附物与(110)面间的相互作用程度, 采用式(1)、 (2), 对H2O分别在IrO2(110)和RuO2(110)表面各自的12种不同吸附位进行几何优化及能量计算. 将H2O以最大成键度置于吸附点表面, 计算H2O在(110)表面的运动趋势. 计算结果表明, 仅在两种吸附方式下, H2O向(110)表面运动, 而其它吸附方式则使H2O背离(110)表面运动, 如图2所示, 即均在不饱和五配位的Mecus原子上发生H2O吸附, 且H2O的取向分别为垂直表面且氢原子向上(记为Mecus-up)和氢原子平行表面(记为Mecus-par)两种.
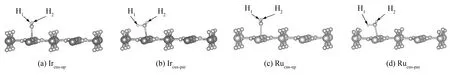
图2 (110)面吸附水分子驰豫后的Slab模型Fig.2 Slab model of (110) relaxation after H2O adsorption
表1为H2O在IrO2(110)和RuO2(110)表面吸附后的结构参数和吸附能结果. 发现H2O发生吸附后分子中的键角增大, 键长增长, 说明因Ircus或Rucus的吸附, H2O中的原子间相互作用减小, H2O的活性有所增加. 表明IrO2与RuO2均能有效促使H2O发生解离, 特别是IrO2, 使H2O键角增幅、 键长增加更多, 能更有效促进H2O的解离.
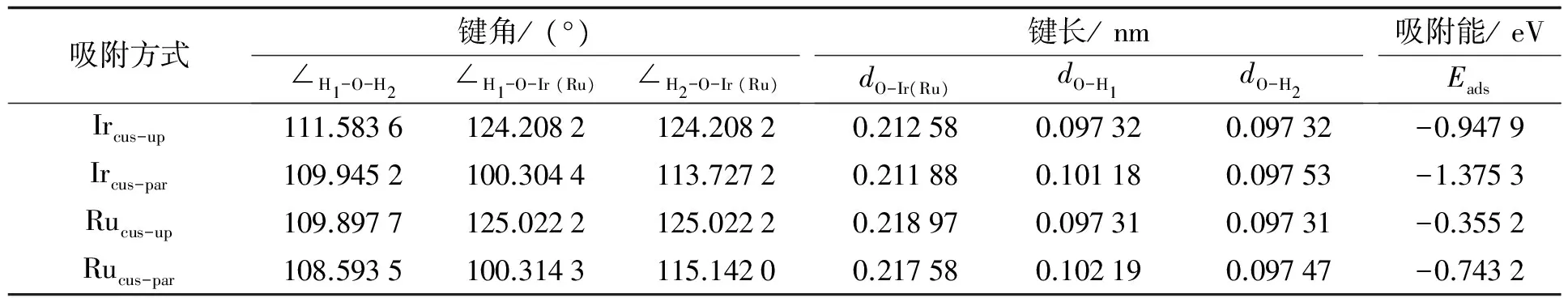
表1 H2O在IrO2(110)、 RuO2(110)表面吸附后的结构参数和吸附能
以Mecus-up方式发生吸附时, H2O整体仍垂直于(110)表面, 如图2 (a)、 2(c)所示, 虽较吸附前H2O键角增大, 键长增长, 但H1、 H2与O的关系变化基本同步, 因此, 可将此吸附方式归为物理吸附. 而当H2O以Mecus-par的方式被吸附后, H2O与(110)面有一定的夹角, 如图2(b)、 2(d)所示, 即此时H2O的H原子不再平行于(110)表面, 而是H1-O-Me键角在100 °左右, H2-O-Me键角则在113° ~116 °之间; 另外, H原子有脱离(110)表面的趋势, 且由于O-H1键长大于O-H2键, O-H1伴有断裂趋势, 结果造成H2O分解, 生成H1和OH2, H1则与(110)表面最邻近的O原子结合形成新的羟基, 而H2则通过原H2O中的O原子吸附于Mecus原子上. 这一吸附过程主要由于O原子与表面Mecus原子间电子转移引起的, 故认为在此处的吸附方式为解离吸附.
一般认为, 键长越短, 吸附能越低, 则原子间相互作用就越强, 即彼此间的吸附作用也就相对较强. 从计算结果(见表1)可知, Mecus-par-O键长均短于Mecus-up-O键, 且Mecus-par方式的吸附能低于Mecus-up吸附能, 说明H2O更容易以Mecus-par的方式吸附于 (110)面, 而且此时H2O有分解倾向.
另外, 由表1还可发现, Ir和Ru对H2O的作用存在明显差异: H2O与Mecus相互作用后, (110)面上的Mecus-up-O和Mecus-par-O键长均为IrO2(110) < RuO2(110), 吸附能亦如此, 说明H2O与IrO2(110)面相互作用强于与RuO2(110)面的相互作用. 这从另一个角度说明IrO2是比RuO2更有效的H2O分解催化剂.
2.2 电子态密度(DOS)
图3为H2O分别以Ircus-up和Ircus-par两种方式, 在IrO2(110)表面发生吸附前后, Slab模型中最外3层原子的电子态密度, 同时图中还给出了最外层Obr原子、 吸附位Ircus原子和H2O的分波态密度(垂直虚线为费米能级处). 图4为H2O在RuO2(110)表面以Rucus-up和Rucus-par方式吸附前后, 表面Slab模型最外3层原子的电子态密度, 以及最外层Obr原子、 吸附位Rucus原子和H2O的分波态密度.

图3 H2O平行和垂直方式吸附IrO2(110)的电子态密度(DOS) Fig.3 DOS of IrO2(110) by absorption of H2O perpendicular/paralleling to (110) surface
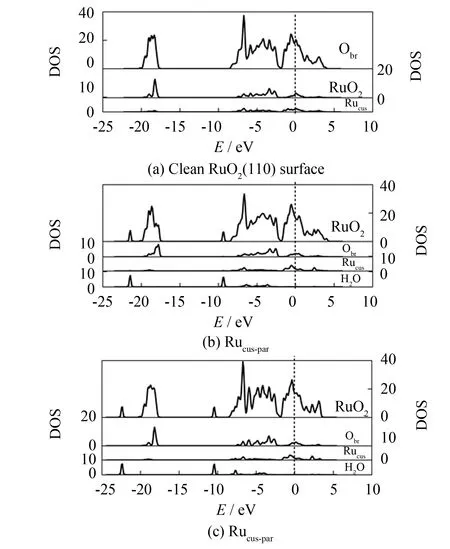
图4 H2O平行和垂直方式吸附RuO2(110)的电子态密度(DOS)Fig.4 DOS of RuO2(110) by absorption of H2O perpendicular/paralleling to (110) surface
从图3(a)中可看出, 在-17.8 eV处出现了尖峰, 这是由Obr表面悬键产生的表面电子态, 与文献[15]工作的结果一致. 与吸附前相比, 吸附后Obr悬键所产生的电子态强度稍有减弱, 表明H2O的表面吸附弱化了(110)面悬挂Obr的作用, 详见图3(b)、 3(c)所示.
H2O的电子态密度主要分布于价带区域, 其费米能级附近只有少量电子分布. Ircus-par时H2O分波态密度的下价带峰出现在-22 eV和-10 eV处, 有别于Ircus-up时位于-23 eV和-11 eV处的尖峰, 这主要是由于H2O的不同吸附取向导致的电子分布差异引起的; 另外, Ircus-par吸附方式使H2O分波态密度的下价带尖峰更靠近费米能级, 表明H2O与(110)面Ircus发生了更强的相互作用, 即H2O更易于吸附于Ircus-par.
从图4 (a)可见, RuO2(110)洁净表面没有出现明显最外层Obr电子态峰, 这是最外层Obr与Ru原子的电子态重合所致. 与H2O吸附后, RuO2(110)的表面电子态密度分布如图4(b)、 4(c)所示, 与IrO2吸附水相似, H2O被吸附后, 其电子态主要占据于价带, 但Rucus-par吸附时, H2O的下价带峰出现在-21.4 eV和-9.3 eV处, 明显高于Rucus-up下的-22.4 eV和-10.4 eV处的尖峰. 且与Rucus-up相比, 发生Rucus-par吸附时, H2O的态密度向高能级区移动幅度相对较大, 更靠近费米能级, 说明此时H2O与RuO2(110)间的相互作用更强烈. 与H2O吸附后, (110)的表面上最外层Obr处的表面电子态峰变得较为平缓, 且费米能级处的电子态强度减弱, 减弱程度Rucus-par大于Rucus-up, 说明Rucus-par吸附时, H2O对RuO2(110)的影响较大.
对比图3、 4还可发现, H2O被(110)面吸附后, Mecus费米能级附近的电子态虽均移向价带, 说明吸附位原子均获得电荷, 表现在Ircus导带尖峰由1 eV位置移至2 eV处, Rucus则出现在2.5 eV, 这是H2O中O与(110)面上的Mecus发生吸附所致. 另外, 此时Rucus导带峰位较Ircus更远离费米能级, 说明H2O与Ircus的吸附作用强于与Rucus的吸附作用. 与H2O吸附后, Rucus的电子态强度变化不如Ircus显著, 也说明IrO2(110) 受H2O影响比RuO2(110)大, IrO2(110)与H2O存在更强相互作用, 表明H2O更易于在IrO2(110)表面发生吸附.
综上所述, 根据电子结构的变化, 可以认为在(110)表面, H2O更易于以Mecus-par的方式发生吸附, 且IrO2比RuO2能更有效地将H2O吸附于(110)表面, 这与上文结果是一致的.
2.3 Bader电荷
由上述电子结构分析可知, H2O在IrO2和RuO2(110)表面吸附后, H2O与(110)表面的Mecus原子之间会发生电荷转移, 体系的电荷分布发生改变. 为考察具体的电荷分布变化情况, 本文分析了吸附前后H2O中的Bader电荷布居数, 结果见表2.

表2 H2O在IrO2(110)和RuO2(110)表面吸附前后的Bader电荷布居
由表2可知, 吸附前H2O为电中性, H2O中的O呈氧化态, 电荷量为-1.200 2 e; H呈还原态, 电荷量为0.600 1 e. H2O发生Ircus-par和Ircus-up吸附后, 分别失去0.158 0 e和0.132 1 e, 而发生Rucus-par和Rucus-up吸附时, 分别失去0.102 2 e和0.092 5 e. 被吸附的H2O带有正电, 即H2O将部分电子转移给了吸附位的Mecus原子, 致使(110)表面导电性增强. 相比Rucus, Ircus能由H2O处获得更多电荷, 也从一个侧面表明IrO2比RuO2更易促使H2O发生分解.
由表2还可发现, H2O 发生Mecus-up吸附时丢失电子的能力弱于Mecus-par吸附时, 且在发生Mecus-up吸附时, H2O中两个H失电子水平相同. 而发生Mecus-par吸附时, H1失去电子明显多于H2, 又一次说明Mecus-par吸附H2O后, 能促使H2O的分解, 也表明此时H2O与(110)面的之间的吸附作用也愈强烈.
3 结语
1) H2O在IrO2(110)和RuO2(110)的吸附方式仅两种, 均发生于Mecus处, 本研究从材料计算的角度证实了Lobo等[12]的实验结果. 并进一步阐明吸附细节, 即H2O分别为以垂直表面且氢原子向上Mecus-up和平行表面Mecus-par的方式发生吸附, 同时Mecus-par吸附强于Mecus-up.
2) H2O吸附于(110)后, 其活性均有不同程度的增强; Mecus-par吸附时, H2O有分解趋势. Mecus-up吸附则为物理吸附.
3) 与RuO2(110)相比, IrO2(110) 吸附H2O的作用更强烈.
4) IrO2(110)和RuO2(110)吸附H2O后, H2O向(110)提供少量电子, (110)电导增大.
参考文献:
[1] TRASATTI S, LODI G. Electrodes of conductive metallic oxides part A[M]. Amsterdam: Elsevier Scientific Pulishing Company, 1981.
[3] LERVIK I A, TSYPKIN M, OWE L E,etal. Electronic structure vs. electrocatalytic activity of iridium oxide[J]. Journal of Electroanalytical Chemistry, 2010, 645(2): 135-142.
[4] CORNELL A, HÅKANSSON B, LINDBERGH G. Ruthenium based DSA®in chlorate electrolysis-critical anode potential and reaction kinetics[J]. Electrochimica Acta, 2003, 48(5): 473-481.
[5] SIRACUSANO S, BAGLIO V, STASSI A,etal. Investigation of IrO2electrocatalysts prepared by a sulfite-couplex route for the O2evolution reaction in solid polymer electrolyte water electrolyzers[J]. International Journal of Hydrogen Energy, 2011, 36(13): 7822-7831.
[6] DE PAULI C P, TRASATTI S. Composite materials for electrocatalysis of O2evolution: IrO2+SnO2in acid solution[J]. Journal of Electroanalytical Chemistry, 2002, 538(2): 145-151.
[7] SALIS M, RICCI P C, CAPPELLETTI G,etal. Phonon confinement effect in mixed Sn-Ir oxide nanocrystals[J]. Chemical Physics Letters, 2010, 496(496): 109-112.
[8] SHAO Y Q, YI Z Y, HE C,etal. Effects of annealing temperature on the structure and capacitive performance of nanoscale Ti/IrO2-ZrO2electrodes[J]. Journal of the American Ceramic Society, 2015, 98(5): 1485-1492.
[9] SHAO Y Q, YI Z Y, LOU C Y,etal. Capacitive characterization of Ti/IrO2-SnO2-CeO2electrodes[J]. Chinese Journal of Nonferrous Metals, 2014, 24(10): 2553-2558.
[10] BAGLIO V, D'URSO C, DI-BLASI A,etal. Investigation of IrO2/Pt electrocatalysts in unitized regenerative fuel cells[J]. International Journal of Electrochemistry, 2011(1): 1-5.
[11] WANG Y R, YANG Y F, HU X,etal. Electrochemical performance of Ru-doped LiFePO4/C cathode material for lithium-ion batteries[J]. Journal of Alloys and Compounds, 2009, 481(1/2): 590-594.
[12] LOBO A, CONARD H. Interaction of H2O with the RuO2(110) surface studied by HREELS and TDS[J]. Surface Science, 2003, 523(3): 279-286.
[13] OVER H, KIM Y D, SEITSONEN A P,etal. Atomic-scale structure and catalytic reactivity of the RuO2(110) surface[J]. Science, 2000, 287(5457): 1474-1476.
[14] OVER H. Surface chemistry of ruthenium dioxide in heterogeneous catalysis and electrocatalysis: from fundamental to applied research[J]. Chemical Reviews, 2012, 112(6): 3356-3426.
[15] 念保峰, 敬熠平, 王欣. IrO2(110)表面弛豫行为与电子结构的第一性原理研究[J]. 材料热处理学报, 2016, 37(10): 20-24.
[16] KIM Y D, SEITSONEN A P, OVER H. Adsorption characteristics of CO and N2on RuO2(110)[J]. Physical Review B, 2001, 63(11): 115419.
[17] WENDT S, SEITSONEN A P, KIM Y D,etal. Complex redox chemistry on the RuO2(110) surface: experiment and theory[J]. Surface Science, 2002, 505(1/2/3): 137-152.
[18] GONG X Q, LIU Z P, RAVAL R,etal. A systematic study of CO oxidation on metals and metal oxides: density functional theory calculations[J]. Journal of the American Chemical Society, 2004, 126(1): 8-9.
[19] FANG Y H, LIU Z P. Mechanism and Tafel lines of electro-oxidation of water to oxygen on RuO2(110)[J]. Journal of the American Chemical Society, 2010, 132(51): 18214-18222.
[20] HANSEN H A, MAN I C, STUDT F,etal. Electrochemical chlorine evolution at rutile oxide (110) surfaces[J]. Physical Chemistry Chemical Physics, 2010, 12(1): 283-290.
[21] WANG C C, YANG Y J, JIANG J C. DFT Study of NHx(x=1~3) adsorption on RuO2(110) surfaces[J]. The Journal of Physical Chemistry C, 2009, 113(7): 2816-2821.
[22] WANG C C, SIAO S S, JIANG J C. Density functional theory study of NHx(x=0-3) and N2adsorption on IrO2(110) surfaces[J]. The Journal of Physical Chemistry C, 2010, 114(43): 18588-18593.
[23] WANG C C, YANG Y J, JIANG J C,etal. Density functional theory study of the oxidation of ammonia on RuO2(110) surface[J]. The Journal of Physical Chemistry C, 2009, 113(40): 17411-17417.
[24] WANG C C, SIAO S S, JIANG J C. C-H bond activation of methane via σ-d interaction on the IrO2(110) surface: density functional theory study[J]. The Journal of Physical Chemistry C, 2012, 116(10): 6367-6370.
[25] 薛严冰, 唐祯安, 孙伟民. 水分子在SnO2(110)表面吸附特性的密度泛函计算[J]. 大连交通大学学报, 2012, 33(5): 93-96.
