Structural confirmation of sulconazole sulfoxide as the primary degradation product of sulconazole nitrate
2018-04-17QunXuAshrafKhanDiGaoKristieAdamsFatkhullaTadjimukhamedovShaneTanJohnSimpson
Qun Xu,Ashraf Khan,Di Gao,Kristie M.Adams,Fatkhulla Tadjimukhamedov,Shane Tan,John T.Simpson
Compendial Development Laboratory,United States Pharmacopeia,Rockville,MD 20852,USA
1.Introduction
Sulconazole,1-[2,4-dichloro-β-[(4-chlorobenzyl)thio]phenethyl]-imidazole,is an azole antifungal drug belonging to the family of the imidazole class,and is typically used as a topical antifungal cream or solution for the treatment of dermatomycoses,pityriasis versicolor,and cutaneous candidiasis[1].Sulconazole nitrate was among the drug substances that were undergoing United States Pharmacopeia(USP)monograph modernization[2].The USP sulconazole nitrate monograph describes an HPLC procedure for assay,but lacks any organic impurity procedures[3].Sulconazole nitrate is currently not included in other major pharmacopeias.
A literature search indicated that limited information is available regarding the stability and chromatographic analysis of this drug substance.HPLC methods have been reported for the analysis of sulconazole in biological samples or drug products[4–7],but sulconazole related compounds were not included for method development or evaluation.In terms of stability studies,comprehensive degradation studies under various stress conditions have not been reported.Chen et al.[8]investigated the kinetics of oxidation of sulconazole with peracetic acid and hydrogen peroxide;sulconazole sulfoxide and sulfone were assumed to be the oxidation products.Iwasawa et al.[9]reported the pharmacological studies of the metabolites of sulconazole and highlighted the liability of the molecule towards oxidative conditions.However,no structural information was disclosed in either of those papers.
Oxidation of organic sulfides(thioethers)to sulfoxides is an important degradation and metabolic pathway for a variety of sulfurcontaining pharmaceutical agents,such as montelukast[10],ranitidine[11],penicillin[12],pergolide[13],and methionine-or cysteine-containing peptides[14].The oxidation process could be complicated as N-or S-oxidation might occur[11],and over-oxidation of a sulfoxide to the corresponding sulfone is frequently a competing reaction[15].To gain a better understanding of the stability of sulconazole nitrate and incorporate the impurity profile into USP monographs,it is critical to differentiate the oxidation pathways and conf i rm the structures of the degradation products[16].
Here we report the structural determination of a major degradation product of sulconazole nitrate,sulconazole sulfoxide,which was generated under oxidative conditions.In addition,this material was subsequently employed as an impurity reference for compendial procedure development and validation.The monograph modernization efforts have eventually culminated in an off i cial compendial procedure and a newly introduced impurity,sulconazole related compound A[17,18].
2.Material and methods
2.1.Chemicals and reagents
The drug substance sulconazole nitrate was obtained from the USP Reference Standard.Commercial samples of sulconazole nitrate were purchased from Sigma-Aldrich(St.Louis,MI,USA),Spectrum Chemical(New Brunswick,NJ,USA),Glentham Life Sciences(Corsham,UK),and Erregierre(Sovere,Italy).Hydrochloric acid(37%),glacial acetic acid(≥99.7%),sodium hydroxide solution(10 N,J.T.Baker),trifluoroacetic acid(TFA,99.5%,Acros Organics),ammonium acetate(LC/MS),methanol(LC/MS),and acetonitrile(HPLC and LC/MS)were purchased from Fisher Scientific(Waltham,MA,USA).Hydrogen peroxide(~30%)was obtained from Sigma-Aldrich.Water was purif i ed with a Milli-Q plus system from Millipore(Bedford,MA,USA).
2.2.Instrument and analytical parameters
2.2.1.UHPLC/HPLC
UHPLC–UV analyses were performed on an Agilent Infinity 1290 UHPLC system(Santa Clara,CA,USA).Separation was carried out on an Acquity BEH C18column(2.1 mm × 100 mm,1.7 μm)from Waters(Milford,MA,USA)using a mobile phase system consisting of mobile phase A:methanol and ammonium acetate buffer(pH 5.0;0.1 mM)(20:80,v/v);and mobile phase B:methanol and acetonitrile(40:60,v/v).Analyses were performed at ambient temperature with a flow rate of 0.5 mL/min and a gradient elution varied according to the following program:0 min,40%B;8min,90%B;8.1 min,40%B;and 10 min,40%B.UV detection wavelength was set at 230 nm.The injection volume was 5μL.
HPLC–UV analyses were performed on a Waters Alliance 2695 HPLC system using a Waters Atlantis C18column(4.6 mm×150 mm,5 μm)and a mobile phase system consisting of 0.05%TFA in water and acetonitrile.Separations were achieved at ambient temperature with a f l ow rate of 1.0 mL/min and an isocratic program of 0.05%TFA in water and acetonitrile(50:50,v/v).UV detection wavelength was set at 210nm.The injection volume was 10μL.
Preparative chromatography was performed on a Waters semipreparative HPLC system consisting of a 2335 quaternary gradient module,a 2998 photodiode array detector,a 2707 autosampler,and a Waters fraction collector III.Separations were carried out on an Agilent Prep-C18column(20.1 mm × 150 mm,5μm)using a mobile phase system consisting of 0.05%TFA in water and acetonitrile(65:35,v/v).The f l ow rate was 10.0 mL/min and the run time was 13 min.UV detection was performed at 210nm.The injection volume was 0.4 mL.Fractions were collected based on time mode.
Waters Empower 2 for chromatography software was used for instrument operation control and data acquisition and processing.
2.2.2.NMR analysis
NMR experiments were recorded using a Bruker Avance III NMR Spectrometer(Billerica,MA,USA)with operating frequencies of 600.13 MHz(1H)and 150.90 MHz(13C).Samples were dissolved in DMSO-d6and all NMR spectra were recorded at 25°C.Chemical shifts were reported in ppm down field from tetramethylsilane(TMS,δ=0)as the internal reference.
2.2.3.LC–MS/MS analysis
The LC–MS/MS was performed on an Agilent 6540 UHD Accurate-Mass QTOF LC–MS using an Agilent 1290 UHPLC as an inlet with UV detection at 210nm prior to the mass spectrometer.Data acquisition and analysis were performed using Agilent MassHunter software.ESI was used in positive ion mode.Except for the injection volume(2μL),LC conditions were the same as those used for UHPLC–UV analysis in Section 2.2.1.The mass spectrometer was tuned in 2 GHz extended dynamic range mode(24,350 FWHM resolution at m/z 1521.9715)for accurate mass analyses.Mass accuracy was less than 0.2 ppm for reference masses across the mass range of m/z 100–1700.The mass range analyzed was m/z 100–1200.
2.3.Solution and sample preparation for UHPLC method
A solution of water and methanol(60:40,v/v)was used as the diluent.A resolution solution was prepared by dissolving sulconazole nitrate and sulconazole sulfoxide in the diluent to give a solution having a concentration of 0.15 mg/mL for sulconazole and 1.5μg/mL for sulconazole sulfoxide.The standard solution was prepared at a concentration of 1.5 μg/mL for sulconazole and sulconazole sulfoxide.The sample solution at a concentration of 1.5 mg/mL was prepared by dissolving a sulconazole nitrate bulk material in the diluent.
3.Results and discussion
3.1.HPLC method development
In light of the structural similarities between sulconazole and econazole(another azole antifungal drug),the mobile phases used in the European Pharmacopeia econazole monographs(econazole and econazole nitrate)were directly adopted as a starting point for UHPLC method development[19].Column optimization led to the identification of ethylene bridged hybrid(BEH)C18silica as the choice of stationary phase.The effective chromatographic separation between sulconazole and the major degradant was achieved using a Waters Acquity BEH column(2.1mm×100 mm,1.7 μm)and a mobile phase of methanol-ammonium acetate buffer and methanol-acetonitrile in a gradient elution.In addition to the separation of the major degradant and sulconazole,the method was also capable of separating the major degradant from a minor secondary degradant with a resolution of 1.4.
An HPLC method was also developed as a guiding method for subsequent preparative HPLC operation(Section 2.2.1).Complete separation was achieved for sulconazole and the major degradant using a Waters Atlantis column and a mobile phase of acetonitrile and 0.05%TFA.Although the method did not provide sufficient resolution for the major degradant and the secondary degradant,our initial scale-up experiments on preparative HPLC revealed that the lack of resolution did not pose a problem for the overall purification.
3.2.Forced degradation of sulconazole nitrate
Forced degradation studies of sulconazole nitrate were performed under thermal,thermal and humidity,photolytic,hydrolytic(acid and base),and oxidative conditions(Fig.1).The degradation was monitored and analyzed using the UHPLC method with UV and MS detection and both detection techniques revealed that degradation of sulconazole only occurred under oxidative conditions(Table S1 and Fig.S1).In addition to the major degradant,a trace amount of secondary degradant was also detected when the stress time was prolonged or the concentration of the oxidant was increased(Fig.2).PDA peak purity testing and LC–MS were performed for the sulconazole peak and no co-elution of impurities was detected.
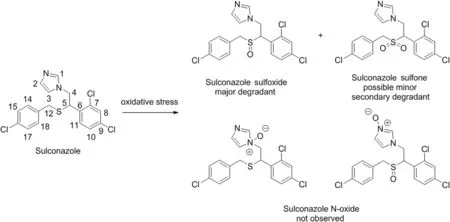
Fig.1.Oxidative stress of sulconazole nitrate.
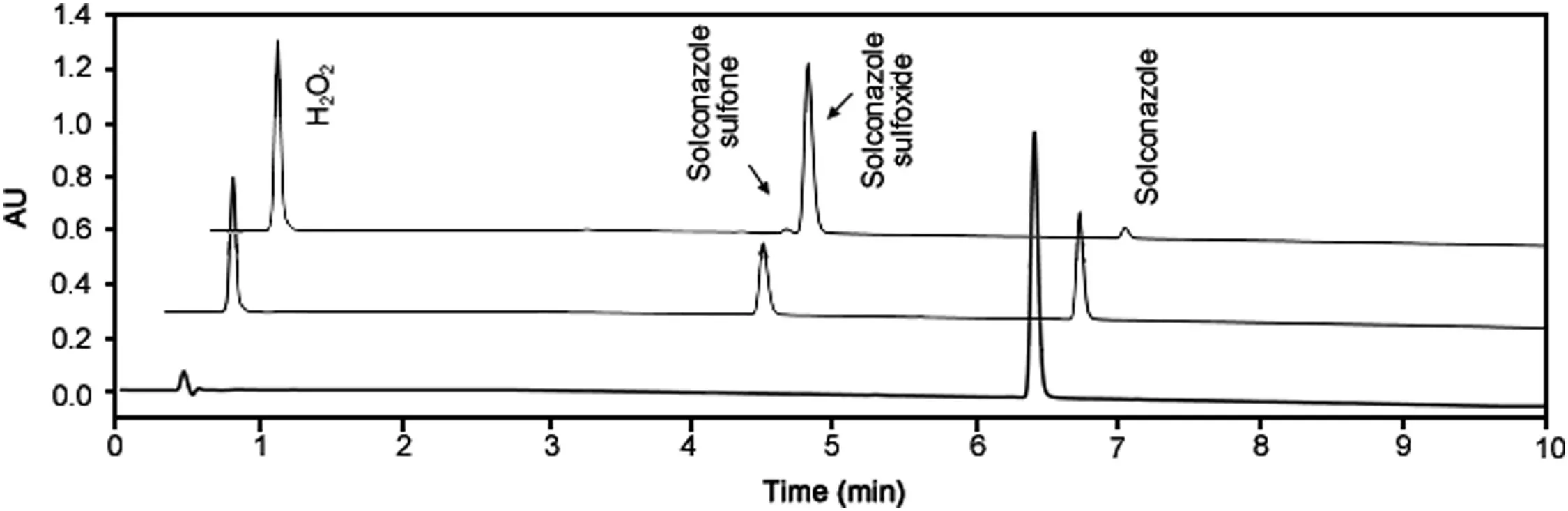
Fig.2.Overlaid chromatograms of oxidative stress of sulconazole nitrate(from bottom to top:chromatogram of sulconazole,chromatograms of sulconazole under oxidative stress for 3 days and 12 days).
The LC–MS analysis showed that the masses of sulconazole and a major oxidation product were m/z 397.0094 and 413.0045,respectively(Fig.S1).The major oxidative degradant showed a protonated molecule that had a delta of 16amu difference from sulconazole,suggesting a single N or S oxidation.A minor ion at m/z 428.9997 with a mass change from sulconazole of 32amu indicated that a secondary degradation,the possible oxidation of sulfoxide to sulfone,or S-oxidation followed by N-oxidation,might occur.
N-oxidation of the imidazole of miconazole nitrate,a similar imidazole antifungal drug,has been reported under hydrogen peroxide-mediated oxidation conditions[20].It is well-documented that both N-and S-oxidation could be competing pathways for drug substances containing both N and S sites susceptible to oxidation,such as ranitidine[21].More importantly,S-oxidation is not always inherently favored over N-oxidation[22].For instance,zofenopril was recently demonstrated to undergo N-oxidation selectively using hydrogen peroxide as an oxidant[23].On the basis of these precedents,the possibility of N-oxide products of sulconazole cannot be excluded at this stage.
3.3.Synthesis and preparative isolation of the degradation product
The scale-up oxidation experiment was performed in a concentrated sample solution using 30%hydrogen peroxide.Complete conversion of sulconazole nitrate to the major degradant was achieved within 20 h(Fig.S2).The experiment was halted when an over-oxidation started to occur.As the reaction proceeded to 100%conversion,the challenge of the purification step was the removal of the minor degradant,which had a retention time close to that of the major peak.
The injection and loading amount were carefully evaluated on an analytical column(Waters Atlantis C18,4.6 mm×150 mm,5μm)to ensure the sufficient separation of the two peaks on the similar preparative column(Prep-C18,20.1 mm × 150 mm,5 μm)while maintaining separation efficiency.To this end,the heartcutting technique was applied and multiple injections of the crude reaction mixture at an appropriate concentration were performed(Fig.S3).The isolated compound was re-analyzed on both analytical HPLC and UHPLC to confirm the identity and purity(Fig.S4).The degradant was isolated as a colorless oil in 93%yield and 98%purity based on HPLC.
3.4.Structural characterization of the major degradation product
3.4.1.NMR studies of sulconazole and sulconazole sulfoxide
Samples of sulconazole nitrate and the isolated major degradation product were subjected to NMR spectroscopic analyses including one-and two-dimensional NMR such as gradient-selected correlation spectroscopy(gCOSY),heteronuclear singlequantum coherence(HSQC)and heteronuclear multiple bond correlation(HMBC)experiments.The NMR spectroscopic data were consistent with the proposed structures of sulconazole and sulconazole sulfoxide(Supplementary material).The1H and13C assignments were made based on characteristic chemical shifts(1D NMR)and were supported by2D NMR data.The1H and13C NMR data and proposed assignments for the two compounds are presented in Table 1 for comparison.
Direct assignments for protons of the two substituted aromatic rings were difficult due to the high degree of similarity inthe1H and13C chemical shifts.In the1H NMR spectrum,a significant chemical shift was observed for methine 5 and methylene 12 protons(Table 1).The change of the splitting pattern indicated the corresponding conformational change after the oxidation of the sulfur[24].More drastic down field shifts of methine 5 and methylene 12 were detected in the13C NMR.In contrast,negligible changes of chemical shifts(1H and13C)on the imidazole ring were observed,indicating that the N-oxidation of imidazole did not occur.Notably,a slight down field shift was detected for methylene 4 in the1H NMR,and a slight upf i eld shift was observed for the same methylene in the13C NMR.These results were in good agreement with NMR data for alkyl groups in a beta orientation to sulfur.The HMBC data showed connections between H1 and C2 and C3,H4 and C5,and H5 and C12(Fig.3).The1H and13C NMR data,in combination with the LC–MS results,provided reasonable evidence in support of the S-oxidation.However,the NMR data alone were not conclusive to differentiate sulfoxide from sulfone,the potential over-oxidation byproduct,as the influence of aliphatic sulfoxide and sulfone groups on the chemical shifts of theα,β,and γ substitutes is very similar[25,26].
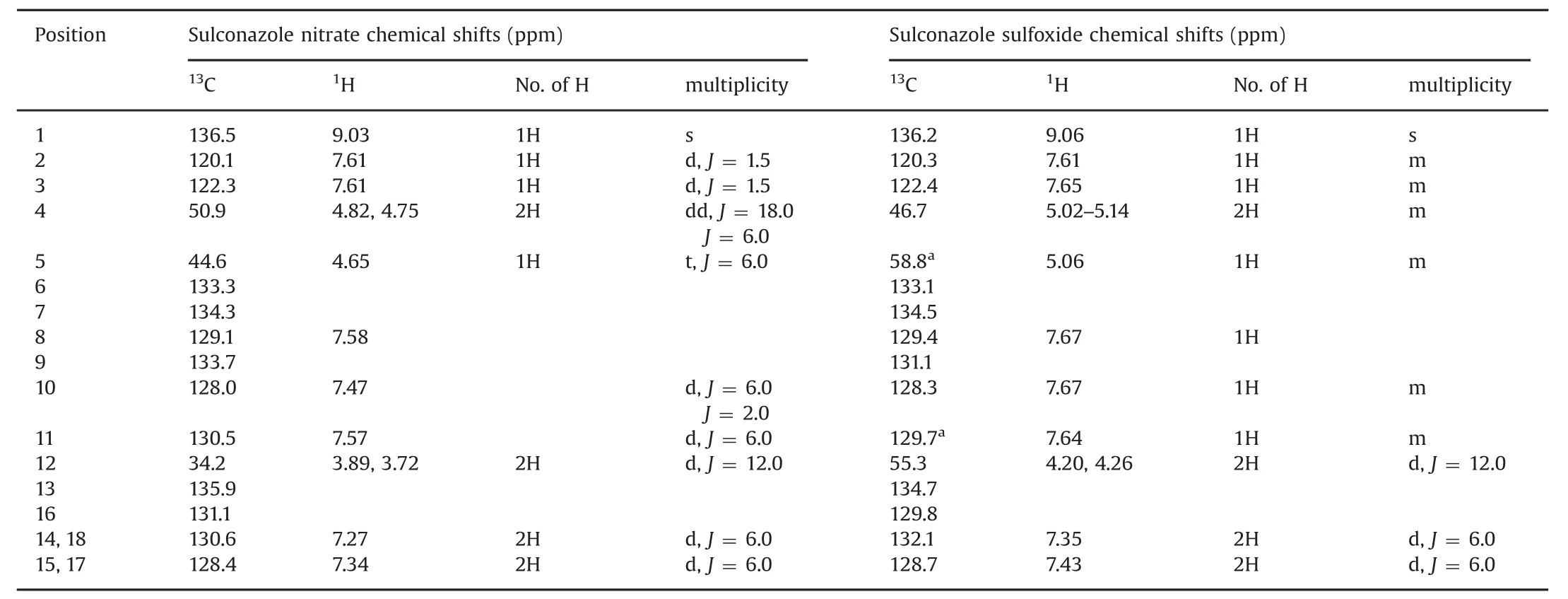
Table 1 Comparative1H NMR and13C NMR data of sulconazole nitrate and sulconazole sulfoxide(DMSO-d6,25°C).
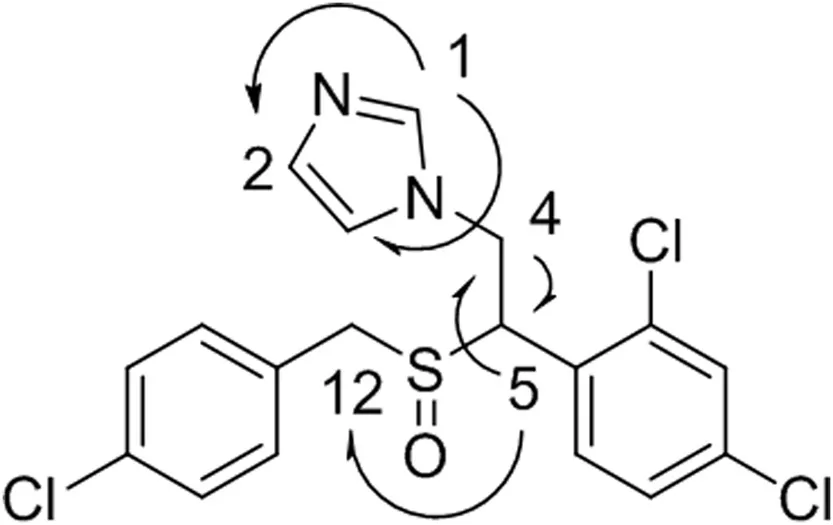
Fig.3.Key1H–13C HMBC correlations of sulconazole sulfoxide.Correlations are shown with arrows.The numbering system is based on the NMR assignments presented in Table 1.
3.4.2.LC–MS/MS studies
The LC–MS/MS analysis was performed using the same UHPLC method(Section 2.2.1).The presence of multiple chlorine atoms in the molecules allows for more precise determination of the fragment ions based not only on the exact mass but also the isotope abundance ratio.The LC–MS spectrum of the oxidative degradant of sulconazole showed a protonated molecule[M+H]+at m/z 413.0048(Fig S5),suggesting an elemental composition of C18H16Cl3N2OS,which is consistent with the structure resulting from S-or N-oxidation.The MS/MS spectrum and possible fragmentation pathways are shown in Fig.4.The MS/MS fragmentation of the[M+H]+ion exhibited a diagnostic fragment ion at m/z239.0136(elemental composition of C11H9N2Cl2),which can be derived from the syn-elimination of alkyl sulfenic acid via a 5-membered cyclic transition state.This McLafferty-type fragmentation has been revealed as a dominant fragmentation pathway for peptides containing oxidized methionine or cysteine residues[27,28].In addition,it was verified as the most facile fragmentation for the styryl-and alkyl-propenyl sulfoxides by deuterium labeling studies[29].Mechanistically,for sulfur-containing molecules this fragmentation only occurs for sulfoxides with aβ-hydrogen atom,and it was indeed observed in numerous reported cases[30–33].The McLaffertytype fragmentation might be a characteristic fragmentation pathway for sulfoxides with aβ-hydrogen atom and could also be used here to distinguish the sulfoxide from the sulfone.Initial examination of the isotopic pattern of this fragment ion indicated that there may be overlapping ion distributions,and a possible fragment b at m/z 240.0212 resulting from a proposed sulfinyl radical cleavage may be contributing to the overall isotopic pattern(Fig.4).Another major fragment at m/z 171.9841(d)may be derived from fragment b via loss of the imidazole ring.The fragment c at m/z 205.0527 was also generated via a McLafferty-type fragmentation after dechlorination of the[M+H]+ion[34–36].In addition to major fragments,the ions at m/z 125.0147(C7H6Cl,e)and 287.9879(C11H10Cl2N2OS,f),generated from cleavage of S-CH2bond were also observed,but were much less abundant(0.05%and 0.02%,respectively),suggesting that this disassociation pathway is not favored.
The MS/MS results of sulconazole nitrate were significantly different from those of sulconazole sulfoxide(Fig.S6).As expected,McLafferty-type fragmentation corresponding to the cleavage of S–CH bond was not observed.The ion g at m/z 125.0158 is predominant under similar activation conditions,suggesting that the cleavage of S–CH2bond is the favored dissociation process resulting in the stable carbocation(Fig.S7).Another main fragment ion j at m/z 183.0030 was generated as a result of the consecutive loss of imidazole and chlorobenzene,which also led to the formation of a stable benzyl thiirane methyl carbocation j.In addition,the intermediate ion h at m/z 328.9717 resulting from the loss of imidazole was also detected.
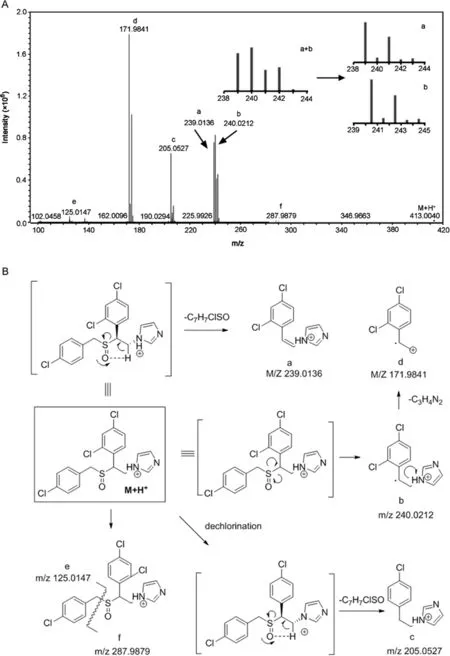
Fig.4.(A)MS/MS spectrum of protonated molecular ion[M+H]+at m/z 413.0048 of sulconazole sulfoxide.(B)proposed fragmentation pathway.The isotope pattern of ion cluster at m/z 239.0136 matches the results of superimposition of two species of a and b as shown in the inserted frame.
As shown in Table 2,all fragmentation assignments were supported by accurate mass data.The accuracy for the masses of these ions,determined relative to the values calculated from their assigned structures,was within 5ppm error.
The comparative MS/MS studies provided compelling evidence that the oxidation occurred on the sulfur atom only,and that the sulfoxide was the primary degradant.Sulconazole sulfone is most likely the product of over-oxidation.Moreover,previous investigations on N-oxides indicated that the mass spectrum of an N-oxide is in general similar to that of parent compound[20],which is also in agreement with our conclusion in support of S-oxidation.
3.5.UHPLC method validation
With the sulconazole sulfoxide in hand,the UHPLC method was validated for specif i city,linearity,accuracy,precision androbustness.System suitability was verified by determining the%RSD of six injections of the standard solution.Retention time,tailing factor and resolution between the sulconazole and sulconazole sulfoxide were also determined(Table S2).The specif i city of the method was established by determining peak purity of sulconazole nitrate subjected to a series of stressed conditions using photodiode array detection.Linearity of the detector responses was determined at five concentration levels of sulconazole nitrate and sulconazole sulfoxide covering 0.75,1.125,1.50,1.875 and 2.25 μg/mL for each compound.To study method accuracy,recovery experiments were carried out by applying the standard addition method.Known quantities of sulconazole sulfoxide of 0.75,1.50 and 2.25 μg/mL—corresponding to 50%,100%and 150%impurity levels—were spiked into a 1.5 mg/mL sulconazole nitrate solution.Accuracy was expressed as the percentage of sulfoxide recovered by the HPLC analysis.Repeatability was studied by carrying out method precision,and determined from results of six independent injections of the 100%spiked solutions.Method robustness was evaluated by analyzing five replicate injections of the robustness solution,sulconazole nitrate(150 μg/mL)and sulconazole sulfoxide(1.5 μg/mL),under each of the varied conditions including temperatures,flow rates,buffer pH,mobile phase B initial compositions,mobile phase B final compositions,and different columns on different instruments.
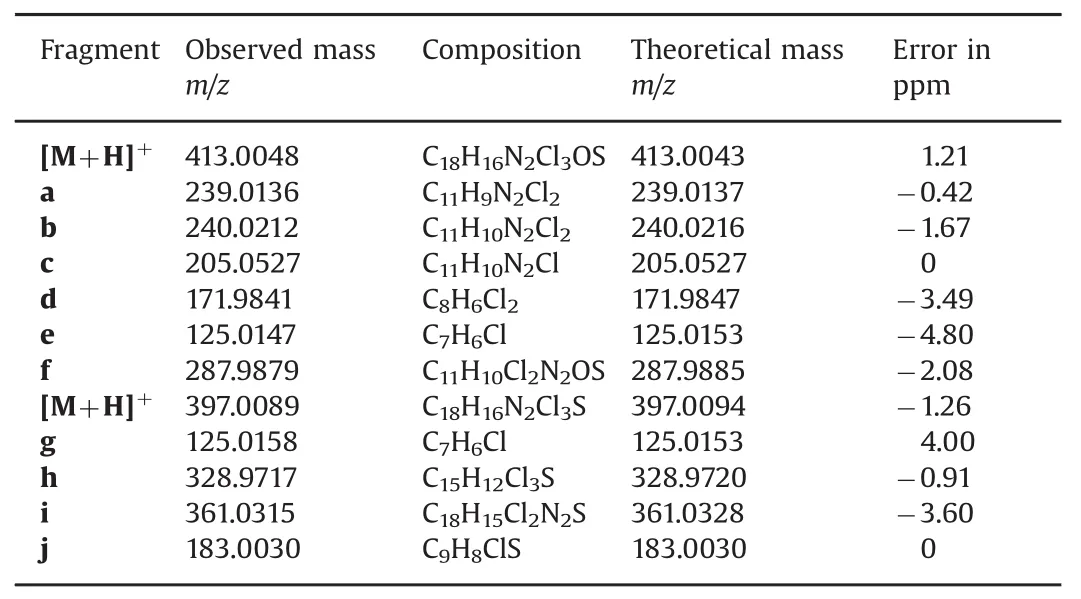
Table 2 Accurate mass measurement of the molecular ions observed from the MS/MS data of sulconazole sulfoxide and sulconazole.The MS/MS spectrum of sulconazole nitrate and proposed fragmentation pathway are provided in Figs.S6 and S7.
The validation results are summarized in Table S3.The results demonstrated that the method is specif i c as sulconazole sulfoxide was separated from sulconazole and no interfering peak appeared at the retention time of the two peaks.Linear responses over the range of concentrations of 0.75–2.25 μg/mL were observed for both sulconazole and sulconazole sulfoxide(Fig.S8).Spike recoveries close to 100%at different levels indicated an acceptable accuracy of the method(Table S4).The low values of%RSD for method precision indicate that the method is reproducible.Finally,robustness studies showed that variations of the operating parameters did not significantly change the chromatographic profiles or the result,indicating that the method was robust under the experimental conditions(Table S5).
4.Conclusions
In summary,a systematic UHPLC study was conducted on sulconazole nitrate.The study included stress testing of sulconazole nitrate,preparative HPLC isolation of the major degradation product,structural confirmation of the degradant,and UHPLC method validation.Based on NMR and LC–MS/MS analysis,the S-oxidation of sulconazole to sulconazole sulfoxide was confirmed as the predominant degradation pathway.In addition,the generated degradation product was utilized as a reference standard for organic impurity HPLC method development and validation.This work displays a typical approach used to support USP monograph modernization.
Conflicts of interest
The authors declare that there are no conflicts of interest.
We thank USP colleagues,Doug M.Podolsky,James R.Austgen,Praveen K.Pabba,Marcela Nefliu,and Sitaram Bhavaraju for help with preparing the manuscript.
Disclaimer
Certain commercial equipment,instruments,vendors,or materials may be identified in this manuscript to specify adequately the experimental procedure(s).Such identification does not imply approval,endorsement,or certification by USP of a particular brand or product,nor does it imply that the equipment,instrument,vendor,or material is necessarily the best available for the purpose or that any other brand or product was judged to be unsatisfactory or inadequate.All product names,logos,and brands are property of their respective owners.
Appendix A.Supplementary material
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.jpha.2017.12.007.
[1]R.A.Fromtling,Overview of medically important antifungal azole derivatives,Clin.Microbiol.Rev.1(1988)187–217.
[2]L.M.Santos,B.Davani,C.M.Anthony,et al.,USP monograph modernization initiative,Am.Pharm.Rev.18(2)(2015).
[3]USP35–NF30 monograph,Sulconazole Nitrate,2012.
[4]M.Fass,B.Zaro,M.Chaplin,et al.,Reversed-phase high-pressure liquid chromatographic analysis of sulconazole in plasma,J.Pharm.Sci.70(1981)1338–1340.
[5]H.Y.Aboul-Enein,I.Ali,Comparison of the chiral resolution of econazole,miconazole,and sulconazole by HPLC using normal-phase amylose CSPs,Fresenius J.Anal.Chem.370(2001)951–955.
[6]H.Y.Aboul-Enein,I.Ali,Comparative study of the enantiomeric resolution of chiral antifungal drugs econazole,miconazole and sulconazole by HPLC on various cellulose chiral columns in normal phase mode,J.Pharm.Biomed.Anal.27(2002)441–446.
[7]E.J.Benjamin,D.L.Conley,On-line HPLC method for clean-up and analysis of hydrocortisone and sulconazole nitrate in a cream,Int.J.Pharm.13(1983)205–217.
[8]S.H.Chen,R.A.Kenley,J.S.Winterle,Sulconazole reactions with peracetic acid and hydrogen peroxide,Int.J.Pharm.72(1991)89–96.
[9]Y.Iwasawa,H.Tamaki,I.Yamaguchi,et al.,Pharmacological studies on sulconazole nitrate(RS44872;SCZ)and its metabolites(2):effects on the cardiovascular,the respiratory,the digestive systems,and the renal and other functions,Oyo Yakuri 27(1984)717–727.
[10]M.M.Al Omari,R.M.Zoubi,E.I.Hasan,et al.,Effect of light and heat on the stability of montelukast in solution and in its solid state,J.Pharm.Biomed.Anal.45(2007)465–471.
[11]P.Viňas,N.Campillo,C.López-Erroz,et al.,Use of postcolumn fluorescence derivatization to develop a liquid chromatographic assay for ranitidine and its metabolites in biological f l uids,J.Chromatogr.B:Biomed.Sci.Appl.693(1997)443–449.
[12]P.G.J.Nieuwenhuis,Conversion of penicillins and cephalosporins to 1(S)-sulfoxides,US Patent 5442058,1995 Aug.15.
[13]D.J.Sprankle,E.C.Jensen,Pergolide mesylate,in:H.G.Brittain(Ed.),Analytical Profiles of Drug Substances and Excipients,21,Academic Press,New York,1992:375.
[14]J.M.Froelich,G.E.Reid,Mechanisms for the proton mobility-dependent gasphase fragmentation reactions of S-alkyl cysteine sulfoxide-containing peptide ions,J.Am.Soc.Mass Spectrom.18(2007)1690–1705.
[15]G.Caron,P.Gaillard,P.Carrupt,et al.,Lipophilicity behavior of model and medicinal compounds containing a sulfide,sulfoxide,or sulfone moiety,Helv.Chim.Acta 80(1997)449–462.
[16]ICH,Guidance for Industry Q1A(R2)Stability Testing of New Drug Substances and Products,2003.
[17]Pharmacopeial Forum,41(5)In-Process Revision:Sulconazole Nitrate,2016.
[18]USP40–NF35,Monograph:Sulconazole Nitrate,2017.
[19]European Pharmacopeia,8.4,Econazole Monograph,and Econazole Nitrate Monograph,2015.
[20]T.S.Belal,R.S.Haggag,Gradient HPLC-DAD stability indicating determination of miconazole nitrate and lidocaine hydrochloride in their combined oral gel dosage form,J.Chromatogr.Sci.50(2012)401–409.
[21]USP40–NF35,Ranitidine Hydrochloride,2017.
[22]G.H.Loew,Y.T.Chang,Theoretical studies of the oxidation of N-and S-containing compounds by cytochrome P450,Int.J.Quantum Chem.48(1993)815–826.
[23]T.Ramesh,P.N.Rao,R.N.Rao,LC–MS/MS characterization of forced degradation products of zofenopril,J.Pharm.Biomed.Anal.88(2014)609–616.
[24]H.Shin,H.Song,S.Cho,et al.,The structure of 1-[2-[[(4-chlorophenyl)-methyl]thio]-2-(2,4-dichlorphenyl)ethyl]-1H imidazole(sulconazole)nitrate,C18H16Cl3N3O3S,Bull.Korean Chem.Soc.18(1997)14–18.
[25]P.S.Beauchamp,R.Marquez,A general approach for calculating proton chemical shifts for methyl,methylene and methine protons when there are one or more substituents within three carbons,J.Chem.Educ.74(1997)1483–1484.
[26]R.J.Abraham,J.J.Byrne,L.Griffiths,1H chemical shifts in NMR.Part 27:proton chemical shifts in sulfoxides and sulfones and the magnetic anisotropy,electric field and steric effects of the SO bond,Magn.Reson.Chem.46(2008)667–675.
[27]X.Jiang,J.B.Smith,E.C.Abraham,Identif i cation of a MS-MS fragment diagnostic for methionine sulfoxide,J.Mass Spectrom.31(1996)1309–1310.
[28]H.Steen,M.Mann,Similarity between condensed phase and gas phase chemistry:fragmentation of peptides containing oxidized cysteine residues and its implications for proteomics,J.Am.Soc.Mass Spectrom.12(2001)228–232.
[29]L.K.Liu,C.Y.Su,W.S.Li,Hydrogen rearrangement in alkyl-,styryl-and alkyl propenyl sulfoxides,Org.Mass Spectrom.24(1989)338–342.
[30]R.Smakman,T.J.de Boer,The mass spectra of some aliphatic and alicyclic sulphoxide and sulphones,Org.Mass Spectrom.3(1970)1561–1588.
[31]P.Wright,A.Alex,D.Gibson,et al.,Characterisation of sulphoxides by atmospheric pressure ionization mass spectrometry,Rapid Commun.Mass Spectrom.19(2005)2005–2014.
[32]S.Kern,R.Baumgartner,D.E.Helbling,et al.,A tiered procedure for assessing the formation of biotransformation products of pharmaceuticals and biocides during activated sludge treatment,J.Environ.Monit.12(2010)2100–2111.
[33]G.S.Prasad,S.Girisham,S.M.Reddy,Studies on microbial transformation of albendazole by soil fungi,Indian J.Biotechnol.8(2009)425–429.
[34]K.P.Madhusudanan,V.S.Murthy,D.Fraisse,Dehalogenation reactions in chemical ionization mass spectrometry,J.Chem.Soc.,Perkin Trans.II 9(1989)1255–1260.
[35]H.Budzikiewicz,Reactions between substrate molecules and chemical ionization reagent gases prior to ionization,Org.Mass Spectrom.23(1988)561–565.
[36]D.Volmer,L.Karsten,Mass spectrometric analysis of nitrogen-and phosphorus-containing pesticides by liquid chromatography–mass spectrometry,J.Am.Soc.Mass Spectrom.5(1994)655–675.
杂志排行

Journal of Pharmaceutical Analysis的其它文章
- Advancements in the preparation of high-performance liquid chromatographic organic polymer monoliths for the separation of small-molecule drugs
- Novel degradation products of argatroban:Isolation,synthesis and extensive characterization using NMR and LC-PDA-MS/Q-TOF
- Physicochemical characterization,the Hirshfeld surface,and biological evaluation of two meloxicam compounding pharmacy samples
- Anti-diabetic activity of quercetin extracted from Phyllanthus emblica L.fruit:In silico and in vivo approaches
- Quantitative determination of erlotinib in human serum using competitive enzyme-linked immunosorbent assay
- Ultrasensitive electrochemical determination of metronidazole based on polydopamine/carboxylic multi-walled carbon nanotubes nanocomposites modified GCE