纳米二硫化钼的制备及其蒽加氢性能
2018-04-12刘忠杉王冬娥潘振栋马怀军姜玉霞田志坚
李 敏, 刘忠杉, 王冬娥, 潘振栋, 马怀军, 姜玉霞, 田志坚
(1.中国科学院 大连化学物理研究所 洁净能源国家实验室, 辽宁 大连 116023;2.中国科学院大学, 北京 100049; 3.新疆独山子天利实业总公司, 新疆 独山子 833699)
二硫化钼(MoS2)具有较高的催化活性和抗硫、抗中毒性能,其为油品悬浮床催化加氢过程中较理想的催化剂[1-5]。悬浮床加氢工艺的特点是将催化剂和反应底物及氢气混合一起通过反应器。催化剂悬浮在反应底物中,随着反应底物一次通过反应器无需考虑其失活问题[6]。因此,制备高活性分散型MoS2催化剂已成为目前研究的一个热点。
根据原子配位方式和层间堆积方式不同,MoS2有3种晶体结构,分别为1T-、2H-和3R-型。其中2H-型属于六方晶系,由2个S-Mo-S单元构成的1个中心对称的晶胞,是最稳定的结构。其具有类石墨烯的层状结构,由S-Mo-S的“三明治”层状结构组成,层与层之间由较弱的范德华力相连接,层内Mo、S间由较强的共价键相连接。这种层状结构形成了2种不同的暴露面,分别为沿层间剥离的基面(basal plane)和沿层内Mo-S键剥离的棱面(edge)[7]。基面化学性质比较稳定,而棱面的化学性质比较活泼,可作为催化反应的活性位,这种各向异性使得2H-MoS2的尺寸、形貌对其催化性能的影响十分显著[8-9]。因此,可通过开发新的合成方法,控制其结构及形貌以得到高活性2H-MoS2催化剂。
近年来,关于纳米MoS2制备方法的报道已有很多,如剥离法、高温热解、高温硫化、气相沉积、低温熔盐、水热等方法[10-11]。其中,水热法具有可调变因素多(如原料、反应温度、时间等)、反应条件温和、操作简单等优势,已成为合成特殊形貌纳米MoS2材料常用的一种方法。Xie等[12-13]以七钼酸铵和硫脲为钼源和硫源,采用水热法制备MoS2纳米片,通过调变七钼酸铵和硫脲的摩尔比得到了O原子注入MoS2纳米片;通过提高水热合成温度得到了多缺陷MoS2纳米片。O原子的注入以及含丰富缺陷位的MoS2纳米片在电化学析氢过程中均表现出高的催化活性。Li等[14-15]采用水/溶剂热法,以七钼酸铵和硫脲做钼源和硫源,通过调变表面活性剂、溶剂分别得到球形、花状、空心结构的MoS2催化剂。分析该化学反应过程,钼酸根与硫脲溶解后首先生成四硫代钼酸铵((NH4)2MoS4, ATTM),而后被N2H4·H2O还原生成MoS2纳米片。以氧化态前驱体制备MoS2催化剂,使硫化较为困难和不完全[16],而以硫化态前驱体来制备MoS2催化剂不仅可解决这一问题,而且有效缩短其形成过程。
ATTM常被用做煤液化和重质油加氢热解过程中催化剂的前驱体[17-18]。Derbyshire等[19]用ATTM做前驱体分散于煤的表面或加入到反应混合物中,在反应条件下原位分解生成MoS2催化剂,该催化剂与煤或反应物紧密接触,表现出高的加氢裂化活性。刘大鹏等人[20]先用液相法合成了十六烷基三甲基四硫代钼酸铵,将其做前驱体经高温热解制得了多孔MoS2固体。尽管该方法可得到高活性MoS2催化剂,但反应过程复杂,实验条件比较苛刻,可重复性差。目前对该过程中催化剂的结构及构效关系的研究尚未见报道。
笔者采用钼硫化合物ATTM做单一前驱体,采用水热法制备2H-MoS2催化剂,考察了不同种类的表面活性剂对该催化剂结构、形貌的影响,并以蒽催化加氢为模型反应,研究其催化加氢构效关系。
1 实验部分
1.1 原料与试剂
七钼酸铵((NH4)6Mo7O24·4H2O,质量分数99%)、乙醇(C2H5OH,质量分数99.5%)、浓氨水(NH3·H2O,质量分数25%~28%)、硫化铵((NH4)2S,分析纯)、水合肼(N2H4·H2O,质量分数80%),天津科密欧化学试剂有限公司产品;十六烷基三甲基溴化铵(C19H42NBr,CTAB,质量分数96%)、二-2-乙基己基琥珀酸酯磺酸钠(C20H37NaO7S,AOT)、聚乙烯吡咯烷酮((C6H9NO)n,PVP)、聚乙二醇(HO(CH2CH2O)nH,PEG),国药集团化学试剂有限公司产品;蒽(C14H10,质量分数98%)、十三烷(C13H28,质量分数98%),阿拉丁试剂公司产品。四硫代钼酸铵((NH4)2MoS4,ATTM,质量分数99%)、去离子水自制。
表面活性剂分子的结构式如图1所示。CTAB是一种阳离子表面活性剂,在水溶液中易解离成十六烷基三甲基胺(CTA+)和Br-。AOT是一种阴离子表面活性剂,是由顺丁烯二酸酐与2-异辛醇酯化后再与NaHSO3反应形成的。PEG和PVP属于非离子型表面活性剂,分别由乙二醇和乙烯基吡咯烷酮单体聚合而成。AOT、PEG和PVP均易溶于水。
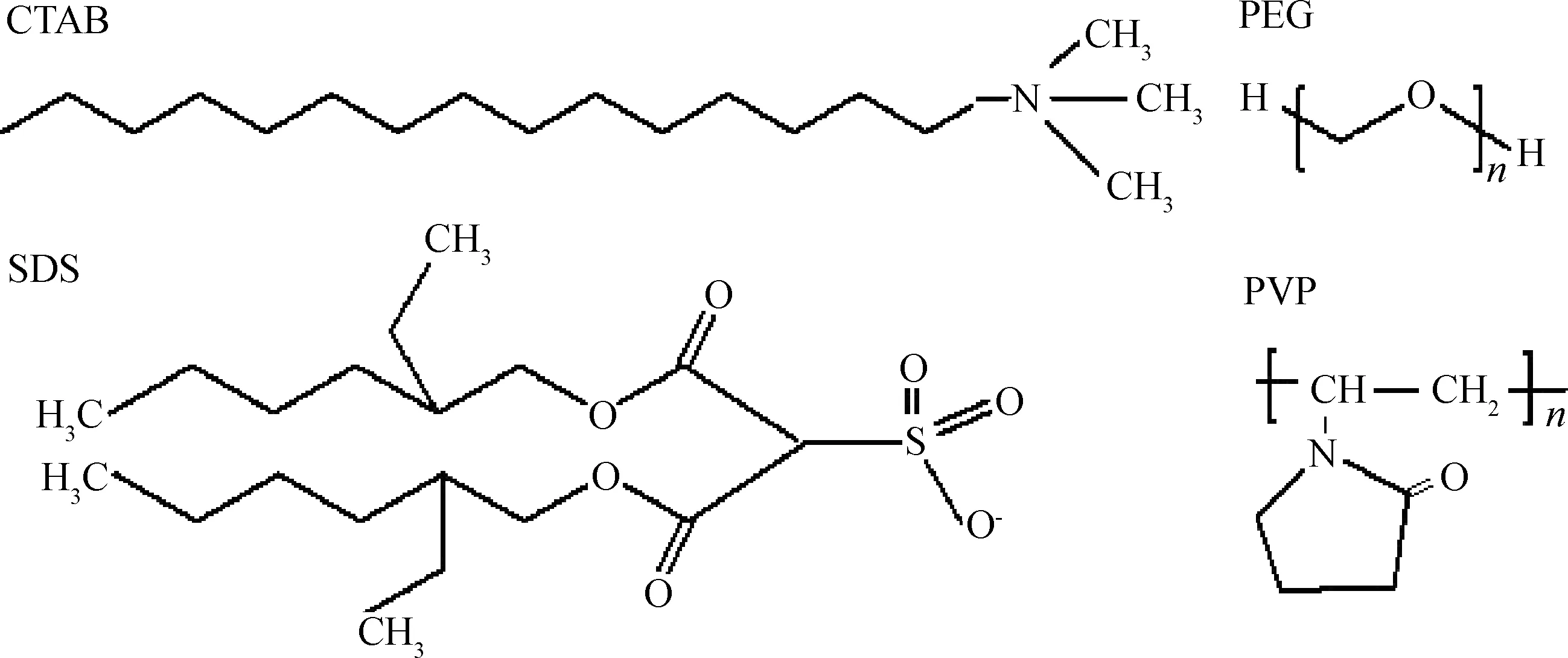
图1 表面活性剂的结构式Fig.1 The structural formulas of CTAB, AOT, PEG and PVP
1.2 ATTM和MoS2的制备
将10.00 g七钼酸铵加入到35 mL浓氨水,搅拌至七钼酸铵晶体完全溶解,加入76.6 g硫化铵溶液,形成深红色溶液,置于60℃恒温水浴中搅拌反应2 h,然后置于冰水浴中冷却、结晶4 h。抽滤,用乙醇洗涤3次,60℃真空干燥4 h,得到ATTM。
将表面活性剂0.40 g CTAB、0.56 g AOT,0.38 g PVP、0.41 g PEG分别溶于70 mL蒸馏水,加入1.42 g的ATTM、2 mL的水合肼N2H4·H2O,混合均匀,置于100 mL聚四氟乙烯内衬高压反应釜中,140℃晶化24 h;冷却、过滤,分别用乙醇和水洗涤3次,70℃真空干燥24 h,得到样品,所得样品分别命名为AT-CTAB、AT-PEG、AT-AOT、AT-PVP。
1.3 MoS2的表征
采用荷兰PANalytical公司X’Pert PROX型X-射线粉末衍射仪进行XRD表征,以CuKα(λ=0.15418 nm)为辐射源,管电压35 kV,管电流40 mA,扫描范围5°~75°,扫描速率5°/min。
采用美国Nicolet公司Nicolet iS50型红外光谱仪进行FT-IR表征。采用美国Micromeritics Instrument公司ASAP2020型物理吸附仪进行BET表征。样品的组分分析在CHNS元素分析仪(CHNS, Thermo Fisher Science公司)和STA449F3型热分析仪(TG,德国NETZSCH公司)上完成;热分析样品质量为5~10 mg,在N2气氛中(气体流速为30 mL/min),从25℃以10℃/min速率升温至860℃,测试范围为:40~860℃。样品的形貌分析在日本JEOL公司JSM7800F型透射电子显微镜上进行,加速电压200 eV。
1.4 MoS2催化剂加氢性能评价
在100 mL不锈钢高压反应釜中进行蒽加氢性能的评价。催化剂0.15 g、蒽3.00 g、溶剂十三烷30.00 g;氢气压力8 MPa,反应温度400℃,反应时间4 h,搅拌速率300 r/min。
采用装有氢离子火焰检测器的Agilent 7890A的气相色谱分析仪(HP-5色谱柱:60 m×320 μm×0.25 μm)对液相产物进行分析。
蒽加氢产物有十四氢蒽(14HN)、八氢蒽(8HN)、四氢蒽(4NH)、二氢蒽(2HN)等。评价悬浮床MoS2催化蒽加氢性能的指标包括加氢产物的选择性、蒽的转化率和蒽的加氢率。其中蒽加氢产物的选择性、蒽的转化率及加氢率以碳平衡法计算,具体计算公式如下:
某一加氢产物的选择性(si):
si=Ai/ATotal×100%
其中Ai、ATotal分别指某一加氢产物的色谱峰面积和所有加氢产物色谱峰面积的总和,即:
ATotal=A2HN+A4HN+A8HN+A14HN
蒽的转化率(x):
x=(nA0-nA)/nA0×100%
其中,nA指反应后蒽物质的量,nA0指反应前蒽物质的量。
加氢率(yH):
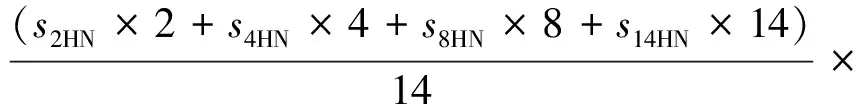
2 结果与讨论
2.1 不同表面活性剂辅助所得MoS2催化剂的结构和组分
图2为不同表面活性剂辅助下水热合成所得MoS2的XRD谱。由图2可知,在XRD高角度范围内(30°~75°),所有样品在33.1°和56.5°均出现了宽而弱的衍射峰,对应于2H-MoS2的(110)和(100)晶面衍射峰。表明所得MoS2样品为六方2H-MoS2相。在低角度范围内(5°~30°),AT-PEG,AT-AOT和AT-CTAB在7.5°左右有1个较弱的衍射峰;而AT-PVP在12°左右有1个较弱的衍射峰。根据布拉格方程式,7.5°和12°左右的衍射峰对应的晶面间距分别为1.17 nm和0.73 nm。1.17 nm和0.73 nm对应样品中相邻2个MoS2的层间距。与标准2H-MoS2的层间距(0.62 nm)相比,所得MoS2样品的层间距较大。根据文献报道[14-15,21],MoS2层间距增大主要是由于杂原子插入导致。不同的表面活性剂辅助所得MoS2样品均为插层结构2H-MoS2。CATB、PEG、AOT辅助水热合成样品的层间距大,PVP辅助水热合成样品的层间距小,由于表面活性剂分子结构不同导致2H-MoS2的层间距不同,这与Wang等[22]的报道类似。
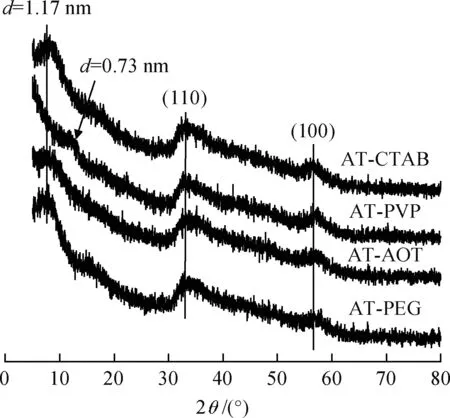
图2 不同表面活性剂辅助水热合成MoS2样品的XRD谱Fig.2 XRD patterns of MoS2 samples preparedwith various surfactants
图3为不同表面活性剂辅助下水热合成所得MoS2样品的FT-IR谱。由图3可知,所有样品均在1400 cm-1处有1个峰,归属为—CH2中C—H的对称振动,来自表面活性剂分子中的—CH2;在1620 cm-1有N—H的特征振动峰[23],来自NH4+。除此之外,AT-PEG和AT-AOT在3300 cm-1左右有1个宽而弱的峰,归属为O—H的伸缩振动峰;在1150 cm-1和1090 cm-1的峰为—C—O—S—的对称和反对称振动峰[22]。AT-CTAB在2830 cm-1和2920 cm-1处有2个振动峰,归属为—CH2的伸缩振动峰[24]。结合表面活性剂的结构式(图1),AT-PEG 和AT-AOT有NH4+的N—H振动峰和表面活性剂分子的—CH2、C—O—S和O—H的振动峰;AT-CTAB和AT-PVP有—NH、—CH2和C—N的振动峰。因此,可推知所得的MoS2样品中含有NH4+和表面活性剂分子。
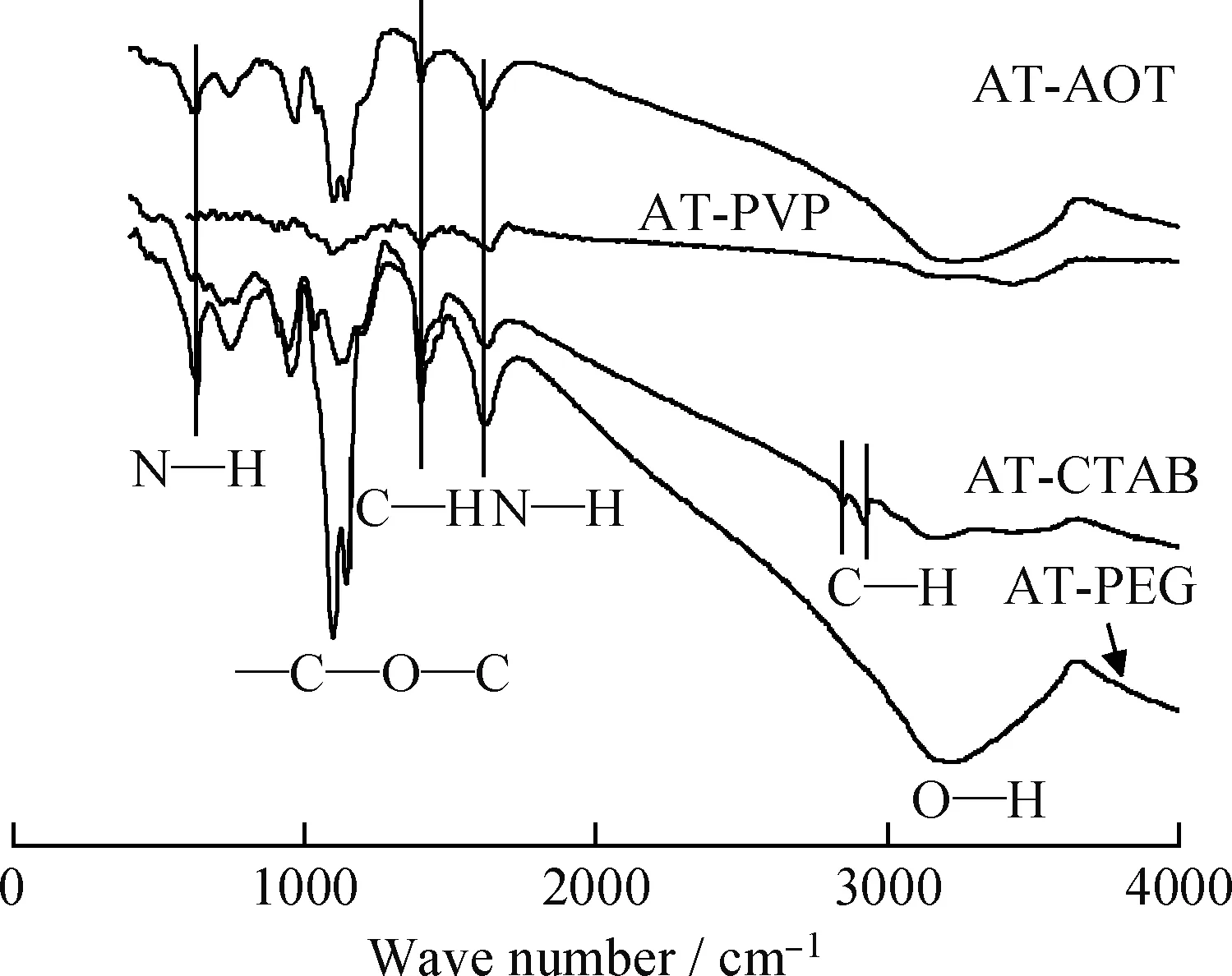
图3 不同表面活性剂辅助水热合成MoS2样品的FT-IR谱Fig.3 FT-IR spectra of MoS2 samples preparedwith various surfactants
表1为不同表面活性剂辅助水热合成MoS2的CHNS元素分析结果。由表1可知,不同表面活性剂辅助水热合成样品中均含有C、H、N元素。C源自表面活性剂分子,N元素则主要源自前驱体ATTM和还原剂水合肼N2H4·H2O分解生成的NH4+。因此,不同表面活性剂辅助所得样品中均包含表面活性剂和NH4+。由n(C)/n(S)可知,AT-PVP和AT-CTAB中n(C)/n(S)的值较高,而AT-PEG和AT-AOT中的n(C)/n(S)值较低,表明AT-PVP和AT-CTAB中包含表面活性剂的量较大,而AT-PEG和AT-AOT中表面活性剂的量较小。
综上所述,MoS2样品为插层结构2H-MoS2,样品中相邻2个MoS2的层间距(1.17 nm或0.73 nm)大于标准的2H-MoS2的层间距(0.62 nm)。增大的层间距是由杂原子的插入导致的,而不是在棱面或是其他位置。由FT-IR和CHNS元素分析结果表明,所得的MoS2样品中包含NH4+和插入的表面活性剂分子。因此,层间插入的杂原子为NH4+和表面活性剂分子。

表1 不同表面活性剂辅助水热合成MoS2的CHNS元素分析结果Table 1 CHNS analysis of MoS2 samples prepared with various surfactants
图4为不同表面活性剂辅助水热合成MoS2样品的TG和DSC曲线。由图4(a)可知,不同表面活性剂辅助下所得MoS2样品的热分解过程相似,样品均有2个质量损失阶段,分别在40~200℃和200~800℃。第1个质量损失阶段可归为H2O脱附,不同的MoS2样品在该阶段的质量损失为5.0%~14.6%;第2个质量损失阶段主要是由NH4+和表面活性剂的脱除所导致,在该阶段样品质量损失变化从大到小的顺序为AT-PVP、AT-CTAB、AT-PEG、AT-AOT。

图4 不同表面活性剂辅助水热合成MoS2样品的TG和DSC曲线Fig.4 TG and DSC curves of MoS2 samples prepared with various surfactants(a) TG; (b) DSC
由图4(b)可知,在280~350℃,AT-AOT、AT-PEG、AT-CTAB和AT-PVP均有1个宽而弱的吸热峰。据报道,NH4+插入的MoS2样品中NH4+的脱附峰在240~340℃[25]。NH4+与MoS2间的相互作用越强,NH4+脱附对应的温度越高。因此,280~350℃的宽峰可能是由NH4+的脱附形成的。在AT-PVP和AT-CTAB的DSC曲线中,在620~640℃有1个宽峰,而AT-PEG和AT-AOT 没有明显的吸热峰。结合TG分析结果,在200~800℃,AT-PVP和AT-CTAB的质量损失大于AT-PEG和AT-AOT的质量损失。在620~640℃,AT-PVP和AT-CTAB中的宽峰可能是由表面活性剂热解所造成[26]。AT-PEG和AT-AOT在该温度范围没有明显吸热峰,可能是因为AT-PEG和AT-AOT 中表面活性剂的量较少。因此,不同表面活性剂在样品中的插入量不同,PVP和CTAB在样品中的插入量较大。PEG和AOT在样品中的插入量较小,这与CHNS的结果一致。据David等[27]报道,新生成的MoS2在水溶液中带一定量负电荷。PVP和CTAB在水溶液中易形成N正离子中心,易吸附到MoS2上;PEG和AOT中的烷氧基及C=O有一定的负电性,不易吸附到MoS2上。因此,表面活性剂插入量的不同可能是由其与MoS2间作用力不同造成的。
综上所述,不同表面活性剂辅助水热合成MoS2样品均为插层结构的2H-MoS2,插入的杂原子为NH4+和表面活性剂。CTAB、PEG、AOT插入的样品层间距较大,PVP插入的层间距较小,层间距的不同可能是由表面活性剂分子结构的不同造成的。不同表面活性剂在MoS2样品中的插入量不同可能是由表面活性剂分子与MoS2间作用力的不同造成的。表面活性剂越易吸附到MoS2上,其在样品中的插入量越大。
2.2 不同表面活性剂辅助所得MoS2催化剂的形貌
采用TEM对不同表面活性剂辅助水热合成MoS2样品形貌进行更直观地表征。图5为不同表面活性剂辅助水热合成MoS2的TEM照片。由图5可知,样品均由MoS2纳米片堆积而成,AT-AOT和AT-PEG中纳米片的堆积密度较大。AT-AOT中纳米片间有团聚,纳米片层数为4~8,长度为10~30 nm。AT-PEG中纳米片团聚现象严重,纳米片层数为3~6,长度为5~20 nm。AT-CTAB和AT-PVP中纳米片的堆积密度较小。AT-CTAB中纳米片排列松散、团聚现象不明显,纳米片层数为2~4,长度为5~20 nm。AT-PVP中纳米片几乎没有团聚,MoS2纳米片层数为1~2,长度为5~10 nm。不同表面活性剂辅助水热合成MoS2样品中,纳米片的尺寸(层数、长度)和排列方式不同。PVP和CTAB的插入量多,样品中纳米片的尺寸小,团聚少;PEG和AOT的插入量少,样品中纳米片的尺寸大,团聚严重。PVP辅助所得样品中纳米片尺寸最小(长度和堆积层数均最小),几乎没有团聚。因此得出,表面活性剂的插入阻碍了纳米片的生长和团聚;表面活性剂的插入量越多,纳米片的尺寸越小,团聚越少。

图5 不同表面活性剂辅助水热合成MoS2的TEM照片Fig.5 TEM images of MoS2 samples prepared with various surfactants(a), (b)AT-AOT; (c), (d)AT-PEG; (e), (f)AT-CTAB; (g), (h)AT-PVP
2.3 不同表面活性剂辅助所得MoS2的催化蒽加氢性能
选用多环芳烃-蒽做模型物,考察MoS2的催化加氢性能。根据Pinilla等[14-15, 27]的报道和笔者之前的工作可知,蒽加氢是一个逐步加氢的过程,首先生成初级加氢产物二氢蒽(2HN),再由2HN加氢生成四氢蒽(4HN),进一步加氢生成八氢蒽(8HN)。深度加氢产物8HN的选择性越高,蒽的加氢率越高,催化剂的加氢性能越好。考察了不同表面活性剂辅助所得MoS2样品的催化蒽加氢性能,结果如表2所示。
不同表面活性剂辅助水热合成MoS2样品做催化剂时,蒽的转化率均很高(为91.5%~98.7%)变化不明显。而蒽的加氢率变化明显,变化从大到小顺序为AT-PVP、AT-CTAB、AT-PEG、AT-AOT。不同表面活性剂辅助水热合成MoS2样品做催化剂时,蒽的加氢产物均由2HN、4HN和8HN组成(见表2)。其中,深度加氢产物8HN选择性变化较明显,其变化趋势与蒽的加氢率变化趋势一致。综上,AT-PVP的加氢活性最好,其次是AT-CTAB。
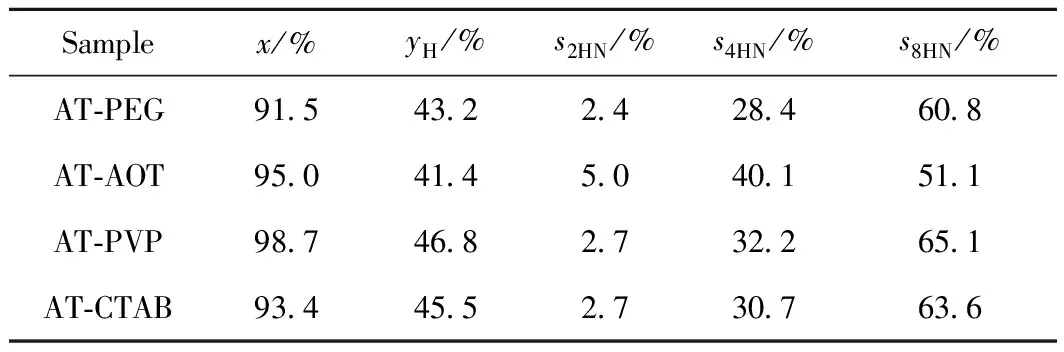
表2 不同表面活性剂辅助水热合成MoS2的催化蒽加氢性能Table 2 The conversion and hydrogenation ratios of AN andthe product selectivity over MoS2 catalysts prepared withdifferent surfactants
Reaction conditions: 3 g anthracene; 30 g tridecane; 0.15 g catalyst;T=400℃;t=4 h; Initial pressure 8 MPa
为了更加明晰MoS2催化剂在加氢过程中的构效关系,将不同表面活性剂辅助水热合成MoS2样品的结构、尺寸、比表面积及催化加氢性能列于表3。由表3可知,与传统催化剂不同,MoS2催化剂的比表面积与MoS2催化蒽加氢活性并无直接相关。这是由MoS2的二维层状结构所决定的,其中催化惰性的基面对比表面积贡献较大,而催化活性的棱边对比表面积的贡献较小。因此,MoS2的催化活性与其比表面积无直接相关。
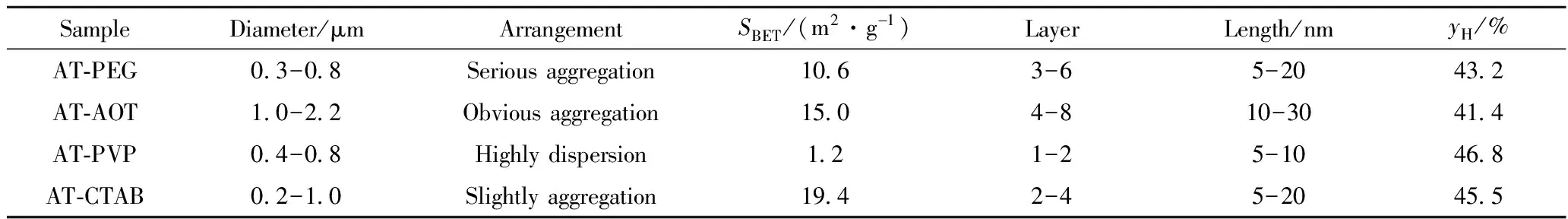
表3 MoS2样品的形貌特征、比表面积及其催化蒽加氢性能Table 3 The morphology characters, surface areas of MoS2 samples and their catalystic performance in anthracene hydrogenation
由表3还可知,AT-PVP中纳米片的尺寸最小,其次是AT-CTAB中纳米片,团聚较少;AT-AOT中纳米片尺寸最大,团聚最严重。MoS2纳米片的尺寸越小(层数、长度),其中棱边位的数量越多,纳米片的团聚越少,越有利于其与反应底物充分接触,进而表现出较高的催化活性。AT-PVP的加氢活性最好,AT-CATB的加氢活性其次,AT-AOT最差。
MoS2催化剂的基面对比表面积贡献较大,较稳定,无催化活性;棱边对比表面积贡献较小,较活泼,是催化反应的活性中心,因此,其加氢活性与其比表面积无关,与其暴露的棱边位数量成正相关。
3 结 论
以ATTM做单一前驱体,采用表面活性剂辅助水热合成纳米MoS2。考察了表面活性剂对其结构、形貌及催化加氢性能的影响。
(1)不同表面活性剂辅助水热合成所得样品均为插层结构2H-MoS2纳米片,插入物为NH4+和表面活性剂。但不同样品中纳米片的层间距、表面活性剂的插入量、纳米片的尺寸、团聚均不同。其中,PVP辅助所得样品中纳米片的层间距最小,PVP的插入量最大,纳米片的尺寸小(片长5~10 nm,堆积层数1~2)、团聚最少。插入的表面活性剂分子结构的不同导致样品中MoS2纳米片的层间距的不同;表面活性剂与MoS2作用力不同导致表面活性剂的插入量不同;表面活性剂的插入量越大,所得纳米片的尺寸越小,团聚越少。
(2)PVP辅助所得MoS2催化剂的加氢活性最高,深度加氢产物8HN的选择性和加氢率分别为65.1%和46.8%。MoS2催化剂的加氢活性与其比表面积无直接相关,与暴露的棱边位的数量成正相关。MoS2纳米片的尺寸越小(长度越小,堆积层数越少),其棱边位的数量越多;纳米片的分散性越好,越有利于棱边位的暴露。因此,通过调变表面活性剂实现了对MoS2纳米片结构、尺寸、团聚的控制,最终得到高活性MoS2催化剂。
[1] COLEMAN J N, LOTYA M, O’NEILL A, et al. Two dimensional nano-sheets produced by liquid exfoliation of layered materials[J]. Science, 2011, 331(4): 568-571.
[2] RAO B G, MATTE H S S R, CHATURBEDY P, et al. Hydrodesulfurization of thiophene over few-layer MoS2covered with cobalt and nickel nanoparticles[J]. Chem Plus Chem, 2013, 78(5): 419-422.
[3] TYE C T, SMITH K J. Hydrodesulfurization of dibenzothiophene over exfoliated MoS2catalyst[J]. Catalysis Today, 2006, 116(4): 461-468.
[4] 刘东, 孔学, 李美玉, 等. 噻吩在硫化态Mo基催化剂表面的吸附行为[J].石油学报(石油化工), 2008, 24(6): 657-662. (LIU Dong, KONG Xue, LI Meiyu, et al. Adsorption of thiophene on the surface of sulfided Mo catalysts[J]. Acta Petrolei Sinica (Petroleum Processing Section), 2008, 24(6): 657-662.)
[5] 柴永明, 南军, 相春娥, 等. 以硫化态前驱物制备的NiMoS/γ-Al2O3催化剂表面活性相HRTEM研究[J].石油学报(石油化工), 2007, 23(3): 20-26. (CHAI Yongming, NAN Jun, XIANG Chune, et al. Investigation of active phase of NiMoS/γ-Al2O3prepared by thiosalt as precursor through HRTEM[J].Acta Petrolei Sinica (Petroleum Processing Section), 2007, 23(3): 20-26.)
[6] 张数义, 邓文安, 刘东, 等. 重质油悬浮床加氢技术新进展[J].炼油技术与工程, 2007, 37(2): 1-6. (ZHANG Shuyi, DENG Wenan, LIU Dong, et al. New development of slurry-bed heavy oil hydrocarcking process[J].Petroleum Refinery Engineering, 2007, 37: 1-6.)
[7] HUANG X, ZENG Z Y, ZHANG H. Metal dichalcogenide nanosheets: Preparation, properties and applications[J].Chemical Society Reviews, 2013, 42(5): 1934-1946.
[8] SONG I, PARK C, CHOI H C. Synthesis and properties of molybdenum disulphide: From bulk to atomic layers[J].RSC Advances, 2015, 5(10): 7495-7514.
[9] BAE J J, JEONGH Y, HAN G H, et al. Thickness-dependent in-plane thermal conductivity of suspended MoS2grown by chemical vapor deposition[J].Nanoscale, 2017, 9(7): 2541-2547.
[10] BACKES C, BERNER N C, CHEN X, et al. Functionalization of liquid-exfoliated two-dimensional 2H-MoS2[J]. Angewandte Chemie, 2015, 54(9): 2638-2642.
[11] ROMERO-RIVERA R, VLLE M D, ALONSO G, et al. Cyclohexene hydrogenation with molybdenum disulfide catalysts prepared by ex situ decomposition of ammonium thiomolybdate cetyltri-methylammonium thiomolybdate mixtures[J]. Catalysis Today, 2008, 130(2-4): 354-360.
[12] XIE J F, ZHANG J J, LI S, et al. Controllable disorder engineering in oxygen-incorporated MoS2ultrathin nanosheets for efficient hydrogen evolution[J]. Journal of the American Chemical Society, 2013, 135(47): 17881-17888.
[13] XIE J F, ZHANG H, LI S, et al. Defect-rich MoS2ultrathin nanosheets with additional active edge sites for enhanced electro catalytic hydrogen evolution[J]. Advanced Materials, 2013, 25(40): 5807-5813.
[14] LI M, WANG D E, LI J H, et al. Facile hydrothermal synthesis of MoS2nano-sheets with controllable structures and enhanced catalytic performance for anthracene hydrogenation[J]. RSC Advances, 2016, 6(75): 71534-71542.
[15] LI M, WANG D E, LI J H, et al. Surfactant-assisted hydrothermally synthesized MoS2samples with controllable morphologies and structures for anthracene hydrogenation[J].Chinese Journal of Catalysis, 2017, 38: 597-606.
[16] HENSEN E J M, BEER V H, VEEN J A R V, et al. A refinement on the notion of type I and II (Co)MoS phases in hydrotreating catalysts[J].Catalysis Letters, 2002, 84(1-2): 59-67.
[17] YANG L, WANG X Z, LIU Y, et al. Layer-dependent catalysis of MoS2/graphene nanoribbon composites for efficient hydrodesulfurization[J].Catalysis Science & Technology, 2017, 7(3): 693-702.
[18] LAI W K, CHEN Z, ZHU J P, et al. A NiMoS flower-like structure with self-assembled nanosheets as high-performance hydrodesulfurization catalysts[J].Nanoscale, 2016, 8(6): 3823-3833.
[19] DERBYSHIRE F. Role of catalysis in Coal Liquefaction research and development[J].Energ & Fuels, 1989, 3(3): 273-277.
[20] 刘大鹏, 赵瑞玉, 柴永明. 十六烷基三甲基四硫代钼酸铵的合成与表征[J].石油学报(石油化工), 2004, 20(6): 28-31. (LIU Dapeng, ZHAO Ruiyu, CHAI Yongming. Synthesis and characterization of cetyltrimethal ammionium tetrathiomolybdates[J].Acta Petrolei Sinica (Petroleum Processing Section), 2004, 20(6): 28-31.)
[21] LI J H, WANG D E, MA H J, et al. Ionic liquid assisted hydrothermal synthesis of MoS2double-shell polyhedral cages with enhanced catalytic hydrogenation activities[J].RSC Advances, 2017, 7(38): 23523-23529.
[22] WANG X F, LI Y J, GUAN Z R X, et al. Guest-host interactions and their impacts on structure and performance of nano-MoS2[J]. Nanoscale, 2015, 7(2): 637-641.
[23] HU L R, REN Y M, YANG H X, et al. Fabrication of 3D hierarchical MoS2/polyaniline and MoS2/C architectures for lithium-ion battery applications[J].ACS Applied Materials & Interfaces, 2014, 6(16): 14644-14652.
[24] GUO Z Y, MA Q X, XUAN Z W, et al. Facile surfactant-assisted synthesis of CTAB-incorporated MoS2ultrathin nanosheets for efficient hydrogen evolution[J].RSC Advances, 2016, 6(20): 16730-16735.
[25] WANG F Z, ZHENG M J, ZHANG B, et al. Ammonia intercalated flower-like MoS2 nanosheet film as electrocatalyst for high efficient and stable hydrogen evolution[J].Scientific Reports, 2016, 6: 31092.
[26] ZHOU K Q, LIU J J, ZENG W R, et al. In situ synthesis, morphology, and fundamental properties of polymer/MoS2nanocomposites[J].Composites Science and Technology, 2015, 107: 120-128.
[27] DAVID L, BHANDAVAT R, SINGH G. MoS2/graphene composite paper for sodiumion battery electrodes[J]. ACS Nano, 2014, 8(2): 1759-1770.
[28] PINILLA J L, GARCIA A B, PHILIPPOT K, et al. Carbon-supported Pd nanoparticles as catalysts for anthracene hydrogenation[J]. Fuel, 2014, 116: 729-735.
