单基因脂代谢异常的研究进展
2018-03-29秦彦文
杨 颂,秦彦文
(首都医科大学附属北京安贞医院 北京市心肺血管疾病研究所, 北京 100029)
血脂异常是动脉粥样硬化性心血管疾病的重要危险因素,人的血脂水平受年龄、饮食、肥胖及其他疾病等因素的影响。除一部分为生活方式不良导致外,大部分原发性血脂异常是由于单一基因或多个基因突变所致,单基因疾病与早发心血管疾病相关性更显著[1]。本文综述了已知的单基因脂代谢异常的临床特点和发病机制。
1 以低密度脂蛋白胆固醇(low-density lipoprotein cholesterol,LDL-C)或总胆固醇(total cholesterol,TC)升高为主的血脂异常
机体内血浆脂蛋白代谢分为内源性、外源性代谢途径及胆固醇的逆转运。内源性代谢途径是指由肝脏合成极低密度脂蛋白胆固醇(very low-density lipoprotein cholesterol, VLDL-C),并转变为中等密度脂蛋白胆固醇和LDL-C,且LDL-C被肝脏或其他器官代谢的过程。该过程的平衡受多种因素影响[2],参与该过程中的酶、蛋白或受体的相关基因变异,可引起以LDL-C或TC升高为主的血脂异常。
1.1 家族性高胆固醇血症(familial hypercholesterolemia,FH)
FH(在线人类孟德尔遗传数据库Online Mendelian Inheritance in Man, OMIM#143890)是由单基因变异引起的最常见的血脂异常。其特点是: LDL-C
水平升高,肌腱黄瘤和/或角膜弓和早发心血管疾病家族史(表1)。FH通常以常染色体显性遗传方式为主,突变携带者比普通人群早发冠心病风险高10~20倍[3];低密度脂蛋白受体(low density lipoprotein receptor,LDLR)基因、载脂蛋白B- 100(apolipoprotein B- 100,ApoB- 100)基因和前蛋白转化酶枯草杆菌蛋白酶9(proprotein convertase subtilisin/kexin type 9,PCSK9)基因突变常导致该病的发生[4]。
LDLR位于染色体19p13.2,该基因突变后使LDLR缺无或功能异常导致LDL颗粒不能被充分清除。表型的严重程度以LDL-C水平为特征,LDL-C水平取决于残余LDLR活性,LDLR突变分为5种类型[5](图1-①~⑤)。
ApoB- 100是LDL颗粒的主要载脂蛋白。由ApoB编码,ApoB位于染色体2p24.1,该基因编码的ApoB- 100对LDL-LDLR复合物的形成至关重要。ApoB突变引起ApoB的结构改变,使LDL与LDLR的亲和力下降约90%[6](图1-⑥)。
PCSK9在肝脏中表达,结合在LDL-LDLR 复合体的LDLR部分,增加LDL和LDLR的亲和力,使内涵体内无法释放LDL,LDLR与LDL被同时降解[7](图1-⑦)。PCSK9位于染色体1p32.3,功能获得性突变增加细胞内LDLR降解导致细胞表面LDLR表达下降,LDL-C从细胞内清除率降低;而PCSK9功能丧失性突变导致相反的表型:细胞表面LDLR数量增加,降低LDL-C水平和较低的心血管疾病风险。
另外,载脂蛋白E(apolipoprotein E,ApoE)基因突变也可引起常染色体显性遗传高胆固醇血症,该类疾病标准筛查应包括ApoE[8- 9]。
除了常染色体显性遗传模式,FH也存在常染色体隐性遗传(ARH;OMIM#603813),是由低密度脂蛋白受体衔接蛋白1基因(low density lipoprotein receptor adaptor protein 1, LDLRAP1)双等位基因失功能突变引起的。LDLRAP1基因位于染色体1p36.11。其参与LDL-LDLR复合体的胞吞作用,突变导致复合体的内化障碍(图1-⑧),从而严重降低LDL的摄取,引起高胆固醇血症。

①无义突变:没有可以检测到的LDLR蛋白;②突变的LDLR无法从内质网运输到高尔基体;③突变的LDLR转运到细胞膜,但无法与LDL-C结合;④突变的LDLR结合LDL-C但无法在被覆小凹内;⑤突变的LDLR不能释放LDL-C,随后不能被细胞膜回收,而在细胞内降解;⑥ApoB失功能突变降低LDL-C颗粒与LDLR连接的能力;⑦PCSK9功能获得性突变明显增加LDL-C和LDLR的亲和力,导致LDL-LDLR复合物被内吞后无法释放LDL;⑧LDLRAP1突变导致复合体的内化障碍
图1FH中基因变异影响胆固醇代谢的类型
Fig1GeneticvariationaffectsthetypeofcholesterolmetabolisminFH
虽然大多数FH是由以上几种基因发生突变导致的,但在约30%的FH患者中没有发现已知突变[5]。本课题组前期对一临床表现为FH的3代家系进行全外显子测序,未发现已知的FH相关变异;而对散发的临床表现为FH的患者进行靶向捕获测序,其中少部分患者也未发现已知致病突变,这说明FH仍有未知遗传变异存在,而FH的发病率也可能因为这些未知因素而被低估。
1.2 胆固醇7-羟化酶基因变异引起的LDL-C异常
胆固醇7-羟化酶仅在肝脏中表达,其在胆汁酸形成的经典途径中启动胆汁酸的合成。由胆固醇7-羟化酶(cholesterol 7-alpha hydroxy-lase,CYP7A1)基因编码(OMIM#118455),该基因位于染色体8q12.1。CYP7A1基因突变引起CYP7A1活性降低,降低了胆固醇分解代谢与胆汁酸的合成,使肝内胆固醇含量增加,肝脏胆固醇水平的增加下调了肝脏的LDLR,导致血浆LDL-C水平增加(表1)。功能缺失突变携带者早发动脉粥样硬化的风险增加。
1.3 溶酶体酸性脂肪酶缺乏症
溶酶体酸性脂肪酶(lysosomal acid lipase,LAL)缺乏症,是一种常染色体隐性遗传的脂肪酶A(lipase A, LIPA)基因突变引起的代谢异常(OMIM#613497),LIPA位于染色体10q23.31。该基因突变引起LAL活性降低导致溶酶体内胆固醇酯和TG积累,细胞内游离胆固醇减少导致内源性胆固醇合成增加。同时,细胞内游离胆固醇减少使HDL-C流出减少,最终表现为血浆LDL-C水平升高、HDL-C水平降低[10]。LAL缺乏的表型取决于剩余的LAL活性(表1)。
2 以三酰甘油(triglyceride,TG)升高为主的血脂异常
通过饮食摄入的胆固醇和TG在小肠中合成乳糜微粒,并接受来自HDL的ApoE和载脂蛋白C(apolipoprotein C,ApoC),成熟后激活脂蛋白脂酶(lipoprotein lipase,LPL),核心的TG被水解,释放游离脂肪酸,产生能量。乳糜微粒残粒在肝脏被分解代谢的过程为外源性代谢途径,该过程涉及的单基因脂代谢异常主要表现为TG升高和混合型血脂异常。
家族性(外源性)高三酰甘油血症又名脂蛋白酶缺乏症或家族性高乳糜微粒血症等。最常见的原因是LPL突变导致LPL缺乏。几乎所有这些突变已被证明是降低或消除LPL活性,导致富含TG的脂蛋白,主要是乳糜微粒的积累[11- 12]。另外,ApoC2编码的ApoC-Ⅱ,存在于HDL、乳糜微粒和VLDL,是LPL激活的一个重要辅助因子,ApoC2突变,影响LPL的激活,从而导致TG蓄积[13]。临床上表现为反复发作胰腺炎,空腹TG常超过11.3 mmol/L(1 000 mg/dL)。
本课题组前期对极端的高TG血症患者进行靶向捕获测序,其中部分患者未携带已知致病突变,但携带有ApoA5等其他相关基因新突变,ApoA5基因簇对高密度脂蛋白胆固醇(high-density lipoprotein cholesterol,HDL-C)影响较大,也有文献报道该基因影响TG水平[14]。

表1 以LDL-C或TC升高为主的血脂异常临床表现Table 1 Clinical presentations of dyslipidemia based on higher LDL-C or TC
3 混合型血脂异常
家族性β脂蛋白血症(FDBL;OMIM # 617347)是一种常染色体隐性遗传的混合型高脂血症。该病主要受ApoE影响,该基因位于染色体19q13.32,主要有3个等位变异,即E2、E3和E4。纯合子的E2变异(ApoE2/E2)可能发展为FDBL。ApoE2基因型携带者从循环排除乳糜微粒和VLDL残余颗粒的能力较低,残余颗粒循环时间较长,残余颗粒的核心与LDL和HDL颗粒的脂质交换使其变为异常胆固醇。这些异常的胆固醇可以导致动脉粥样硬化。FDBL的特点是TC和TG水平升高[15];与早发冠心病风险增加相关[16]。
4 以HDL-C降低为主的血脂异常
HDL在ATP结合盒转运蛋白A1(ATP-binding cassette transporter A1,ABCA1)和载脂蛋白A1(apolipoprotein A1,ApoA1)作用下,在肝脏和小肠形成新生的HDL,并在卵磷脂胆固醇脂酰基转移酶(lecithin-cholesterol acyltransferase,LCAT)的作用下成熟。HDL-C中大部分胆固醇酯通过胆固醇酯转移蛋白(cholesteryl ester transfer protein, CETP)转移给VLDL和LDL,后者进入肝脏被分解代谢。该过程中相关基因变异可引起HDL水平的变化(图2)。
4.1 家族性低α脂蛋白血症
家族性低α脂蛋白血症(OMIM#604091)是一种罕见的常染色体显性遗传血脂紊乱,受ApoA1/ApoC3/ApoA4/ApoA5基因簇编码的脂蛋白影响,该基因簇位于染色体11q23.3。家族性低α脂蛋白血症表型取决于受影响的基因簇(表2)。
4.2 丹吉尔病/家族性HDL缺陷症
丹吉尔病(OMIM#205400)是由ABCA1基因突变引起的常染色体隐性遗传病,该基因位于染色体9q31.1。ABCA1促进胆固醇和磷脂从外周细胞和肝脏流出到脂质颗粒很小的ApoA1(新生HDL)[17](图2)。ABCA1突变的纯合和杂合子患者表型严重程度不同(表2)。
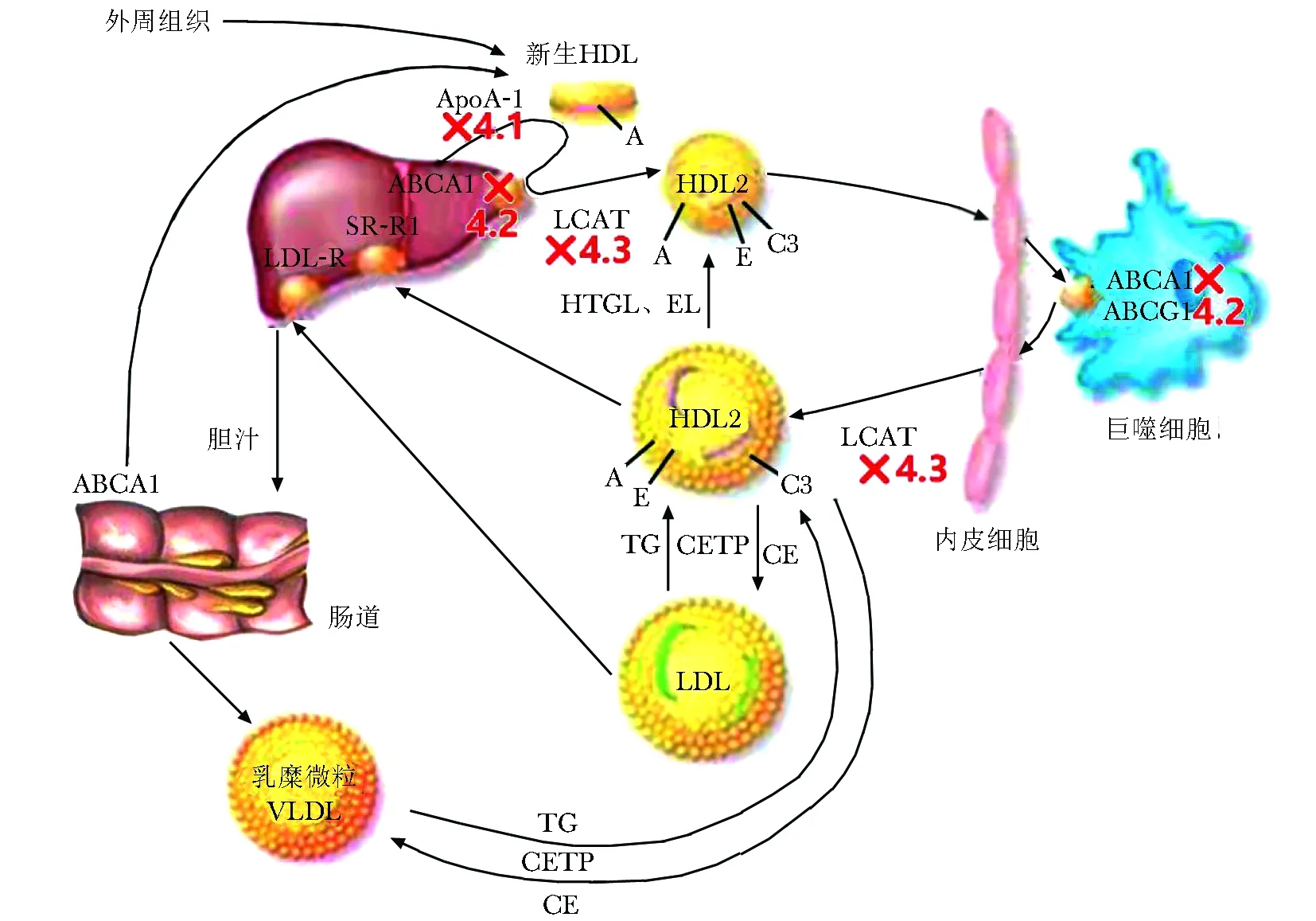
4.1家族性低α脂蛋白血症;4.2丹吉尔病/家族性HDL缺陷症;4.3家族性LCAT缺陷综合征图2 以HDL-C降低为主的血脂异常代谢途径Fig 2 Abnormal metabolic pathway of hyperlipidemia based on HDL-C reduction

疾病基因其他临床表现家族性低α⁃脂蛋白血症 ApoA1/ApoC3/ApoA4/ApoA5基因簇 TG水平降低。脂肪吸收不良。扁平和肌腱黄色瘤、角膜弓和角膜混浊丹吉尔病 ABCA1 TG中度增加,LDL⁃C水平降低。肝脾肿大,扁桃体肿大,轻度角膜混浊,神经病变和早期动脉粥样硬化LCAT缺乏LCAT 家族性LCAT缺乏LDL⁃C水平降低,肾功能不全,角膜混浊和溶血性贫血 鱼眼病角膜混浊
4.3 家族性LCAT缺陷综合征
LCAT缺陷综合征是一种十分罕见的脂代谢紊乱,患者可伴有角膜浑浊,可能呈不完全显性遗传模式,是由于LCAT突变导致的。该基因位于染色体16q22.1,LCAT突变引起LCAT缺乏和不成熟的HDL颗粒的形成(图2)[18]。LCAT突变表现为病情较重的总LCAT缺乏(FLD;OMIM#245900)或病情较轻的鱼眼病(FED;OMIM#136120)(表2)。
5 特殊类型的血脂异常
5.1 家族性高谷固醇血症
谷固醇是一种非胆固醇,在人体内微量存在。健康人中,肠上皮细胞的ABCG5和ABCG8转运蛋白促进多数谷固醇进入肠腔,剩余的谷固醇通过ABCG5和ABCG8转运蛋白作为乳糜微粒转运至肝脏,排泄到胆汁[19]。高谷固醇血症是由于ABCG5和ABCG8突变使肠道谷固醇吸收增加,肝脏排泄减少导致血浆谷固醇升高[20]。谷固醇血症(OMIM#210250)是一种常染色体隐性遗传病,表现为肌腱黄色瘤和早发心血管疾病,症状较FH轻(表3)。
5.2 脑腱黄瘤病
脑腱黄瘤病(CTX;OMIM 213700)是一种罕见的隐性遗传代谢异常,由编码固醇27-羟化酶蛋白的CYP27A1突变导致。CYP27A1位于染色体2q35,它在几乎所有的体细胞的线粒体酶中表达。固醇27-羟化酶参与胆汁酸的形成,该酶缺乏导致胆固醇不能转化为胆汁酸而使血浆胆汁醇和胆固烷醇增加,胆固烷醇在肌腱和大脑中成瘤积累。脑腱黄瘤病的表现多样,可能涉及多个器官(表3)。
5.3 CETP缺陷症
CETP突变导致CETP缺乏, HDL颗粒中的胆固醇酯转运至其他脂蛋白时发生障碍,造成HDL颗粒中胆固醇堆积,HDL-C明显升高,LDL-C降低,纯合子HDL-C>2.6mmol/L;杂合子中度增高;但与早发冠心病未明确相关性。

表3 特殊类型的血脂异常Table 3 Special type of dyslipidemia
6 展望
随着新一代测序技术的发展,已发现越来越多的脂代谢相关基因。在这篇综述中,描述了已确定的脂代谢单基因疾病。但并非所有的遗传因素都已被发现。单基因脂代谢异常的患者通常有更高的早发心血管疾病风险,因此,对单基因脂代谢异常的早期诊断非常重要。目前,由于临床表现的异质性及重叠的临床表现,很难区分具体的单基因疾病,单基因脂代谢异常的发病率是被低估的,而对单基因脂代谢异常的正确诊断有利于未来的精准治疗,例如:对于大多数高胆固醇血症患者(包括FH),他汀类药物治疗是金标准,但谷固醇血症患者使用他汀类药物效果不佳,低谷固醇饮食联合依折麦布治疗更为有效。对部分血脂极端异常和难以降到理想水平的患者,通过基因筛查确诊后,可能为这些患者寻找新的治疗方法提供理论靶点。
针对这些单基因脂代谢异常疾病,北京安贞医院-北京市心肺血管疾病研究所制定了相关的基因panel,对极端血脂患者进行基因变异筛查,通过靶向捕获测序技术,在患者中发现了部分已知的血脂相关单基因致病突变,并对患者的家属也进行了基因的检测,为其治疗和生活方式提供相应的指导。另外,在部分未发现已知变异的患者中,也发现了一些新的基因变异,下一步将对这些基因变异进行相应的功能验证,旨在寻找新的血脂异常的诊断和治疗靶点,也为心血管疾病的早期预防提供新的理论依据。
对于单基因脂代谢异常,未来研究的目标是准确定位所有单基因遗传血脂代谢异常的致病基因,并明确其致病机制。人类遗传学与生物医学研究的整合将为血脂代谢异常疾病提供新的诊治靶点。
[1] Khera AV, Won HH, Peloso GM,etal. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia[J]. J Am Coll Cardiol,2016,67:2578- 2589.
[2] van der Velde AE, Brufau G, Groen AK. Transintestinal cholesterol efflux[J]. Curr Opin Lipidol,2010,21:167- 171.
[3] Nordestgaard BG, Chapman MJ, Humphries SE,etal. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinici-ans to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society[J].Eur Heart J,2013,34:3478- 3490.
[4] Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment[J]. J Clin Invest,2003,111:1795- 1803.
[5] van Schie MC, Jainandunsing S, van Lennep JER. Monogenetic disorders of the cholesterol metabolism and premature cardiovascular disease[J]. Eur J Pharmacol,2017,816:146- 153.
[6] Andersen LH, Miserez AR, Ahmad Z,etal. Familial defective apolipoprotein B- 100: A review[J].J Clin Lipidol,2016,10:1297- 1302.
[7] Durairaj A, Sabates A, Nieves J,etal. Proprotein convertase subtilisin/Kexin type 9 (PCSK9) and its inhibitors: a review of physiology, biology, and clinical data[J]. Curr Treat Options Cardiovasc Med,2017,19:58- 69.
[8] Awan Z, Choi HY, Stitziel N,etal. APOE p.Leu167del mutation in familial hypercholesterolemia[J]. Atherosclerosis,2013,231:218- 222.
[9] Wintjens R, Bozon D, Belabbas K,etal. Global mole-cular analysis and APOE mutations in a cohort of autoso-mal dominant hypercholesterolemia patients in France[J].J Lipid Res,2016,57:482- 491.
[10] Maciejko JJ. Managing cardiovascular risk in lysosomal acid lipase deficiency[J]. Am J Cardiovasc Drugs,2017,17:217- 231.
[11] Johansen CT, Hegele RA. Genetic bases of hypertriglyceridemic phenotypes[J]. Curr Opin Lipidol,2011,22:247- 253.
[12] Chokshi N, Blumenschein SD, Ahmad Z,etal. Geno-type-phenotype relationships in patients with type I hyperlipoproteinemia[J]. J Clin Lipidol,2014,8:287- 295.
[13] Johansen CT, Hegele RA. The complex genetic basis of plasma triglycerides[J]. Curr Atheroscler Rep,2012,14:227- 234.
[14] Cui G, Li Z, Li R,etal. A functional variant in APOA5/A4/C3/A1 gene cluster contributes to elevated triglyc-erides and severity of CAD by interfering with microRNA 3201 binding efficiency[J]. J Am Coll Cardiol,2014,64:267- 277.
[15] Hopkins PN, Brinton EA, and Nanjee MN. Hyperlipoproteinemia type 3: the forgotten phenotype[J]. Curr Atheroscler Rep,2014,16:440- 458.
[16] Mahley RW. Apolipoprotein E: from cardiovascular dise-ase to neurodegenerative disorders[J]. J Mol Med (Berl),2016,94:739- 746.
[17] Rothblat GH, Phillips MC. High-density lipoprotein heterogeneity and function in reverse cholesterol transport[J]. Curr Opin Lipidol,2010,21:229- 238.
[18] Ossoli A, Simonelli S, Vitali C,etal. Role of LCAT in atherosclerosis[J]. J Atheroscler Thromb,2016,23:119- 127.
[19] Yoo EG. Sitosterolemia: a review and update of pathophysiology, clinical spectrum, diagnosis, and management[J]. Ann Pediatr Endocrinol Metab,2016,21:7- 14.
[20] Escola-Gil JC, Quesada H, Julve J,etal. Sitosterolemia: diagnosis, investigation, and management[J].Curr Atheroscler Rep,2014,16:424- 431.
