基于SSR标记的四倍体鸭茅遗传图谱加密
2018-03-24唐露金梦雅黄琳凯张旭赵欣欣张新全
唐露,金梦雅,黄琳凯,张旭,赵欣欣,张新全
基于SSR标记的四倍体鸭茅遗传图谱加密
唐露,金梦雅,黄琳凯,张旭,赵欣欣,张新全
(四川农业大学动物科技学院,成都 611130)
【目的】利用转录组测序开发的EST-SSR标记和鸭茅基因组调研测序开发的基因组SSR(genomic-SSR)标记,对已构建的四倍体鸭茅遗传图谱加密,为定位控制鸭茅重要农艺性状的QTL位点奠定基础。【方法】基于拟测交策略,以“楷模”(高杆、多分蘖、宽叶、早熟)和“01436”(矮秆、少分蘖、细叶、晚熟)作为亲本材料进行杂交,得到一个含有214株鸭茅材料的作图群体,利用亲本和随机选取的5个单株对574对EST-SSR标记和150对Genomic-SSR进行引物筛选,PCR产物经8%非变性聚丙烯酰胺凝胶电泳检测后,将扩增条带清晰、在亲本之间存在差异且子代间存在分离的多态性引物用于亲本及群体扩增。将扩增结果按标记类型统计分析,对于亲本间存在差异的条带,按条带有无(有带计1,无带记0)对DNA扩增产物按进行统计,经卡方检验,将分离比例符合1﹕1(亲本基因型为Aaaa×aaaa或aaaa×Aaaa)和3﹕1(亲本基因型为Aaaa×Aaaa)的标记,用于遗传连锁图谱构建。符合作图要求的标记采用HighMap软件进行遗传图谱构建。【结果】最终筛选出符合要求的EST-SSR引物31对和Genomic-SSR引物17对,引物多态性分别为5.4%和11.3%,总的多态性为6.6%。对鸭茅214个作图群体单株及亲本DNA进行扩增,共得到169个多态性位点,其中EST-SSR101个,Genomic-SSR68个位点。169个标记位点经卡方检验分析表明,有89个标记符合孟德尔分离规律,标记可用率为52.7%,其中呈Aaaa×aaaa或aaaa×Aaaa分离类型的标记有79个,呈Aaaa×Aaaa的有10个,其余80个为偏分离标记。将SSR标记整合以前的标记信息,重新构建了一张包含2 551个标记,覆盖7个连锁群,总长度为758.4 cM的鸭茅高密度遗传图谱。加密后的图谱包含SNP标记4 187个,SSR标记84个,各连锁群标记数在166—709个,每个连锁群的平均标记数为364个,LG1包含最多标记数有709个,LG7标记数最少166个,各连锁群长度在60.28—147.09 cM,标记平均密度为0.19—0.76 cM,总的平均图距由原来的0.37 cM缩至0.3 cM,且由于标记密度的改变,各连锁群上标记分布的位置也发生较大变动。【结论】增加了部分SSR标记后,新构建了一张包含2 551个标记,覆盖7个连锁群总长度为758.4 cM的四倍体鸭茅遗传图谱,总长度增加42.63 cM,平均图距由0.37 cM缩至为0.3 cM。
鸭茅;SSR;遗传图谱加密
0 引言
【研究意义】鸭茅属()隶属于禾本科(Gramineae)[1],全属仅一个种,即鸭茅[2]。鸭茅(L)又名鸡脚草或果园草,鸭茅作为一种多年生冷季型牧草,具有耐荫、耐旱和高产、适口性好等优点,现已成为世界大面积栽培的一种重要优良牧草,可应用于青刈、放牧和干草调制[3-4],特别是在温带地区林下种草和石漠化治理中广泛应用[5-6]。它的育种目标主要包括产量、品质等重要性状[7],而分子标记辅助下的多基因聚合分子育种将是今后突破性饲草选育开拓性研究的领域。其中,高密度遗传图谱构建是开展重要农艺性状的QTL和分子标记辅助育种的重要基础。【前人研究进展】鸭茅为多年生异花授粉植物,具有自交不亲和性和严重自交衰退现象,难以产生自交系,有关鸭茅遗传图谱构建的研究报道相对较少,迄今为止,国内外构建了4张鸭茅遗传图谱。2011年,Song等[8]首次报道了同源四倍体鸭茅的SSR连锁遗传图谱,其中父本的遗传图谱包含24个连锁群,总长度为562 cM,有168个位点,平均图距3.3 cM,母本包含26个连锁群,总长度为745 cM,有227个位点,平均图距为3.3 cM,亲本图谱间同源连锁群共有7个。Xie等[9]则采用SRAP和SSR标记构建了世界上首张二倍体鸭茅遗传图谱,并采用双拟测交策略构建了包含284个单株的F1代作图群体,使用20对AFLP引物和65对SSR引物构建了另一张同源四倍体鸭茅高密度遗传图谱[10],用于定位于抽穗及开花相关的QTL。2016年Zhao等[11]利用2 467个SLAF标记和43个SSR标记的构建了一张高密度鸭茅遗传图谱,总长度715.77 cM,平均图距0.37 cM,该研究为鸭茅重要基因定位、候选基因挖掘及克隆搭建了良好平台。【本研究切入点】目前,利用各种分子标记构建的植物遗传图谱已成为遗传育种的重要工具。简单重复序列(simple sequence repeats,SSR)又叫微卫星DNA,在基因组内广泛分布的高多态性标记,拥有共显性和可重复性等优点[12],广泛应用于品种鉴定[13]、系统发育关系研究[14]、遗传多样性研究[15]、遗传连锁图谱的构建[16]和分子标记辅助育种[17]等领域。Zhao等[11]虽然已经构建了高密度的遗传图谱,但由于标记数目有限,且多为SNP标记,以SSR为主的共显性标记数目相对较少,因此,QTL定位效果,特别是隐性基因控制性状的QTL效果欠佳。【拟解决的关键问题】本研究利用转录组开发的EST-SSR标记和鸭茅基因组调研测序开发的Genomic- SSR标记对Zhao等[11]构建的鸭茅遗传连锁图谱进行加密,旨在获得一张含有更多共显性标记、密度更高的四倍体鸭茅遗传连锁图谱,为鸭茅的进一步重要农艺性状QTL分析奠定基础。
1 材料与方法
1.1 作图亲本及作图群体
试验材料包括母本楷模(高秆、多分蘖、宽叶、早熟)和父本01436(矮秆、少分蘖、细叶、晚熟)及214株作图群体。将母本楷模和父本01436材料杂交,获得F1种子,并于温室发芽,2个月后提取材料DNA,利用SSR分子标记进行杂交种真实性鉴定,将F1代群体材料移栽至四川农业大学雅安(38°8′N, 103°14′E,海拔600—620 m,年均温16.0℃,年均降水量1 015.2 mm,年平均日照时数1 161.5 h,年平均无霜期283 d)试验基地中。选取长势良好且存在差异的F1单株杂交获得包含214个单株的作图群体材料,用于鸭茅遗传图谱的构建[18]。
1.2 鸭茅DNA提取
2016年于四川农业大学雅安草学实验基地取生长良好的鸭茅群体及亲本材料幼嫩叶片,放入塑封袋中加硅胶干燥。依照天根植物基因组DNA提取试剂盒中说明书的步骤提取鸭茅基因组DNA。通过0.8 %琼脂糖凝胶电泳和核酸蛋白质检测仪检测DNA浓度和纯度,合格的样品于-20℃冰箱保存备用。
1.3 SSR标记分析
1.3.1 引物筛选 574对EST-SSR引物[19]和150对Genomic-SSR引物[20]用于亲本及子代间多态性筛选。其中,EST-SSR标记引物为四川农业大学牧草分子育种课题组利用耐热材料宝兴及敏感材料01998通过转录组测序(RNA-seq)开发,Genomic- SSR标记引物为二倍体鸭茅材料2006-1通过基因组调研测序(genome survey sequencing)开发。利用亲本和随机选取的5个群体单株进行引物筛选,PCR产物经SDS-PAGE电泳后,将扩增条带清晰、在亲本之间存在差异且子代间存在分离的多态性引物用于亲本及群体扩增。
1.3.2 SSR扩增 SSR反应体系为10 ng·μL-1的DNA模板3 μL、MIX 7.0 μL(dNTP 240 μmol·L-1、Taq酶1.0 U和Mg2+2.5 mmol·L-1),引物浓度0.4 μmol·L-1。PCR反应程序为94℃ 10 min;94℃ 30 s,58—62℃ 30 s,72℃ 1 min,共30个循环;72℃ 10 min,4℃保存。PCR扩增产物经8%的非变性聚丙烯酰胺凝胶检测,用0.1%的AgNO3进行银染色并在NaOH溶液中显色,凝胶在灯光下用数码相机拍照,保存以供分析。
1.3.3 数据分析与图谱构建 根据SSR分子标记的扩增结果,对于亲本间存在差异的条带,按条带有无(有带计1,无带记0)对DNA扩增产物按进行统计,经卡方检验,将分离比例符合1﹕1(亲本基因型为Aaaa×aaaa或aaaa×Aaaa)和3﹕1(亲本基因型为Aaaa×Aaaa)的标记,用于遗传连锁图谱构建。利用北京百迈客研发的高密度遗传图谱构建软件HighMap[21]进行图谱构建,构图过程包括连锁分群、标记排序、基因型纠错和图谱评估共4部分。采用单连锁聚类算法,设置最大遗传距离为20 cM,将标记整合在连锁群上,构建遗传图谱。
2 结果
2.1 SSR标记多态性及分离比例分析
574对EST-SSR引物和150对Genomic-SSR引物中筛选出48对多态引物(电子附表1),多态性引物为6.6%。48对SSR引物对鸭茅群体材料及亲本扩增得到169个多态位点(表1),其中有57个来自母本楷模,有90个来自父本01436,22个为双亲共有位点。引物扩增的多态性位点在2—4个,平均多态位点为3.52个。
经过引物多态性筛选,构建图谱所用的48对SSR引物中有31对为EST-SSR引物,17对为Genomic-SSR引物(图1)。31对EST-SSR引物共扩增得到101个多态性位点,平均为3.26个,17对Genomic-SSR引物共扩增得到68个多态性位点,平均为4个。根据孟德尔遗传定律,标记的期望分离比一般为1﹕1或者3﹕1。通过卡方检验,发现共有89个SSR标记符合分离比,可用于遗传图谱构建,可用率为52.7%,其中EST-SSR引物可以标记55个,Genomic- SSR引物可用标记34个,可用率分别为54.5%和50%。在符合分离比的89个标记中,呈Aaaa×aaaa或aaaa×Aaaa分离类型的标记有79个,Aaaa×Aaaa的有10个,另外80个标记表现为偏分离,偏分离率为47.3%。
2.2 遗传连锁图谱构建
将SSR标记进行筛选,得到可以使用的标记位点89个,加上以前的标记用于作图的标记总数为2 556个。并通过两两标记之间计算MLOD值[22],过滤掉与原图谱中其他SLAF标签的MLOD值均低于3的标记,共得到上图标记2 551个(表2)。以连锁群为单位,采用HighMap软件分析获得连锁群内Marker的线性排列,并估算相邻Marker间的遗传距离,最终得到包含7个连锁群、总遗传距离为758.4 cM的中性遗传图谱(电子附图1)。其中SNP标记4 187个,SSR标记84个,各连锁群标记数在166—709个,每个连锁群的平均标记数为364个,LG1包含最多标记数有709个,而LG7标记数最少166个。各连锁群长度在60.28—147.09 cM,平均密度为0.19—0.76 cM,其中总的标记平均密度为0.3 cM,其中第4连锁群长度为60.28 cM,所含标记却有326个,平均密度为0.19 cM,所以标记分布较为均匀。
2.3 图谱加密前后对比
2.3.1 图谱长度及密度的变化 图谱加密后,各项特征均较原图有所改变(表3)。Zhao等[8]的原图谱有7个连锁群,2 510个标记位点,总长度715.77 cM。原图谱7个连锁群上的总标记数范围为161—688个,其中第7连锁群标记最少161个,最多的为第1连锁群688个,而SSR标记数为43个,第4、5连锁群上未分布SSR标记,密度在0.19—0.68 cM,平均密度为0.37 cM。加密后的连锁群密度为0.19—0.76 cM,平均密度减少了0.07 cM,缩至0.3 cM,总长度增长到758.4 cM,其中各连锁群SNP标记数目未发生变化,仅SSR标记发生改变,尤其第1连锁群增加了21个,第5连锁群新增3个SSR标记。多数连锁群的长度都发生了改变,如第3连锁群新增标记6个,长度由121.74 cM缩为为116.74 cM,平均密度缩短0.02 cM;第5连锁群新增标记3个,长度由77.98 cM缩短到70.48 cM,平均密度缩短0.04 cM。

表1 SSR引物标记多态性及分离类型

表2 新建遗传图谱各连锁群基本信息
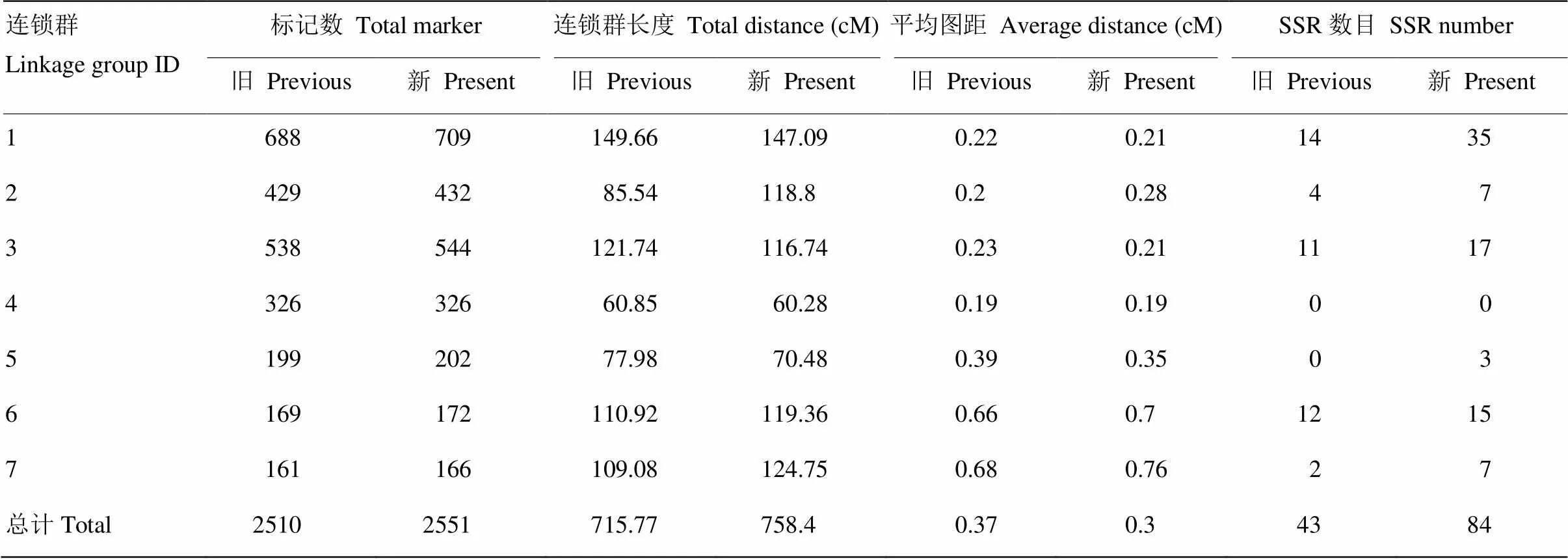
表3 图谱加密前后比对

图1 引物G72对部分鸭茅杂交种的SSR检测图
2.3.2 标记位置的变化 标记位置随着密度变化而变动。比如标记数目变化较大的第1连锁群120—150 cM区间内(电子附表2),旧图谱的标记21个,密度为1.41 cM,新图谱的标记有33个,平均密度缩至0.83 cM。该区间内,新图谱较原图谱新增12个SSR标记,在Marker13902和Marker44715之间新增6个SSR标记,Marker174063和Marker134359之间新增5个,Marker48536和Marker78421新增一个。这些SSR标记的插入导致连锁群内部分标记的位置发生很大的改变,如标记MarkerFOG537N2位于新图谱第1连锁群127.75 cM处,却位于原图谱的起始端0 cM处;位于原图谱13.68 cM处的MarkerFOG21N3,在新图谱中移至132.06 cM处;该区间内原图谱中MarkerB06H11N1与Marker241458,却并没有出现在新图谱120—150 cM区间内。
3 讨论
3.1 作图策略及图谱构建
由于鸭茅为多年生异花授粉植物,具有自交不亲和性和严重自交衰退现象,所以培育诸如DH群体和RIL群体等永久性分离群体在实际中很困难,因此,大部分鸭茅连锁图谱的作图群体大多来自于种内品系间或属内种间杂交产生的F1群体。用此方法杂交所得F1群体进行作图的策略称为“双假测交”(two-way pseudo test cross)。“双假测交”是指2个亲本基因型均为杂合,使其互为测交群体,与测交试验一样,后代基因型的分离比例为1﹕1。双假测交被认为是解决多倍性异花授粉植物遗传连锁图谱构建的有效方法[23-24]。目前已获得的牧草分子连锁图谱大部分所用构图群体均通过双拟测交获得,如冰草((LinnGaertn.)、高丹草×)、多花黑麦草()、多年生黑麦草(L.)等[25-28]。四川农业大学牧草分子育种课题组前期同样采用该方法构建了包含214个单株的作图群体,并构建了基于大量SNP标记的高密度遗传连锁图谱。在此基础上,本文利用通过测序开发的SSR标记,对原图谱进行加密,重新构建了一张包含2 551个标记位点,涉及7个连锁群的高密度鸭茅分子遗传图谱,图谱总长758.4 cM,长度增加42.6 cM,平均图距由0.37缩至为0.3 cM,较加密前,其覆盖度更广,质量更高。但是,由于重新构图时,进行了相关标记的更新以及对相关位点进行了多次排序和纠错,导致许多标记位点间的距离与顺序发生了改变。此外,所有标记位点在连锁群上的分布也并不均匀,而均匀度是衡量图谱质量的一个重要指标。如连锁群1标记位点数高达709个,而连锁群6、7却分别只有172、166个。同时,两种SSR标记在图谱上呈间隔分布,却也出现位点分布聚集化问题,主要分布在第1、3、6连锁群,该结果与xie等[10]结果(1、3、4)较为一致,但笔者的研究结果却显示第4连锁群并未分布SSR标记。造成这些现象的主要原因可能是遗传标记自身的非随机分布(主要分布于染色体的近端或末梢的基因丰盈区域)和这些连锁群遗传差异比较小,缺少多态性标记有关。
3.2 SSR标记的多态性及构图高效性
由于SSR标记具重复性高,特异性强,共显性遗传,操作简单的优点,已逐渐成为基因定位和遗传多样性的首选标记,被广泛应用于分子遗传图谱构建、重要质量或数量性状基因的定位、分子选择辅助育种、指纹图谱构建及亲缘关系鉴定方面[29-30]。但该类标记在基因组内的分布却不是随机的,大部分位于非表达区,而基于转录组开发的EST-SSR标记,主要对编码区扩增,用其构建遗传连锁图,相当于定位功能已知的基因,这将可对决定重要表型的等位基因进行直接鉴定[31]。本研究在574对EST-SSR引物和150对Genomic-SSR引物中筛选出48对多态引物,用于鸭茅遗传图谱的加密,共扩增出169个多态性位点,平均为3.52个,平均多态率为6.63%。由于EST-SSR引物来自较为保守的基因表达区域,很多研究表明genomic-SSR比EST-SSR标记的多态性高[32-33],本研究也得出了类似结论,genomic-SSR标记多态性为11.33%显著高于EST-SSR的5.4%。但总体而言多态性不高,与Xie等的研究结果相比偏低,而与同为四倍体且遗传基础狭窄的陆地棉利用SSR标记进行亲本间引物多态性筛选的结果相差无几(6.8%)[34]。本研究从724对引物中仅仅选择出48对引物(6.63%)在作图群体中分离,这可能和所用亲本材料皆为同源四倍体,只有单剂量类型标记(Aaaa×aaaa、Aaaa×Aaaa)才能在后代群体中呈多态分离且符合作图软件的分离模式,其他标记类型均不能用于作图。在以后的相关研究中,可选择多种标记类型进行分子图谱构建,以增加标记多态性,也可考虑采用多交群体增加亲本之间的遗传差异,使分子图谱增加更多遗传差异信息,提高图谱覆盖率。
4 结论
基于四川农业大学牧草分子育种课题组先前构建的一张鸭茅遗传连锁图谱基础,利用测序开发的EST-SSR及Genomic-SSR标记,以原亲本组合的214个杂交后代为作图群体,增加了部分SSR标记,将2 551个多态性位点绘制在涉及7个连锁群的鸭茅遗传图谱上,图谱总长758.4 cM,长度增加42.63 cM,平均图距由0.37缩至为0.3 cM。
[1] 董宽虎, 沈益新. 饲草生产学. 北京: 中国农业出版社, 2003: 113-117.
DONG K H, SHEN Y X.. Beijing: China Agriculture Press, 2003: 113-117. (in Chinese)
[2] 谢文刚, 张新全, 马啸, 彭燕, 黄琳凯. 鸭茅种质遗传变异及亲缘关系的SSR分析. 遗传, 2009, 31(6): 654-662.
XIE W G, ZHANG X Q, MA X, PENG Y, HUANG L K. Genetic variation and relationship in orchardgrass (L.) germplasm detected by SSR markers., 2009, 31(6):654-662. (in Chinese)
[3] JAFAR A, NASERI H. Genetic variation and correlation among yield and quality traits in cocksfoot., 2007, 145: 599-610.
[4] WANG G Z, ZUO F Y, ZENG B, TANG X F. Comparison on seed production of various orchardgrass., 2011(4): 34-38.
[5] VAN SANTEN E, SLEPER D A. Orchardgrass. America. American Society of Agronomy-Crop Science Society of America-Soil Science Society of America. 1996: 503-534.
[6] 张新全. 优质牧草鸭茅种质资源发掘及创新利用研究. 北京: 科学出版社, 2015.
ZHANG X Q.. Beijing: science press, 2015. (in Chinese)
[7] BUSHMAN B S, LARSON S R, TUNA M,WEST M S, HERNANDEZ A G, VULLAGANTI D, GONG G, ROBINS J G, JENSEN K B, THIMMAPURAM J. Orchardgrass (L.) EST and SSR marker development, annotation, and transferability., 2011, 123(1): 119-129.
[8] SONG Y H, LIU F X, ZHU Z F,TAN L B, FU Y C, SUN C Q, CAI H W. Construction of a simple sequence repeat marker-based genetic linkage map in the autotetraploid forage grassL., 2011,57(57):158-167.
[9] XIE W G, ZHANG B Y, ZHANG X Q, LIU W, CHEN Y X, ZHOU H. Identification and genetic analysis of orchardgrass (L.) hybrids by SRAP molecular markers.,2010, 43(16): 3288-3295.
[10] XIE W G, ZHANG X Q, CAI H, HUANG L K, PENG Y, MA X. Genetic maps of SSR and SRAP markers in diploid orchardgrass (L.) using the pseudo-testcross strategy., 2011, 54(3): 212-221.
[11] ZHAO X X, HUANG L K, ZHANG X Q, WANG J P, YAN D F, LI J, TANG L, LI X L, SHI T W. Construction of high-density genetic linkage map and identification of flowering-time QTLs in orchardgrass using SSRs and SLAF-seq., 2016, 6: 29345.
[12] WANG Y, GEORGI L L, ZHEBENTYAYEVA T N, REIGHARD G L, SCORZA R, ABBOTT A G. High-throughput targeted SSR marker development in peach ()., 2002, 45(2): 319-328.
[13] JIANG L F , ZHANG X Q, MA X , HUANG L K, XIE W G, MA Y M, ZHAO Y F. Identification of orchardgrass (L.)cultivars by using simple sequence repeat markers., 2013, 12(12): 5111-5123.
[14] XIE W, ZHANG X, PENG Y. Genetic variation and phylogenetic relationships of orchardgrass(L.) as revealed by SSR markers., 2009, 8: 122-123.
[15] YAN H, ZHANG Y, ZENG B, YIN G, ZHANG X, JI Y, HUANG L, JIANG X, LIU X, PENG Y, MA X, YAN Y. Genetic diversity and association of EST-SSR and SCoT markers with rust traits in orchardgrass (L.)., 2016, 21(1):66.
[16] 张利达, 唐克轩. 植物EST-SSR标记开发及其应用. 基因组学与应用生物学, 2010, 29(3): 534-541.
ZHANG L D, TANG K X. Development of plant EST-SSR markers and its application., 2010, 29(3): 534-541. (in Chinese)
[17] 杨海健, 伊华林. HB柚有性后代的杂种鉴定和遗传多样性分析. 华中农业大学学报, 2012(5): 574-577.
YANG H J, YI H L. Identification and genetic diversity analysis of hybrid progenies from HB pummelo., 2012(5): 574-577. (in Chinese)
[18] Chen C, Sleper D A, Johal G S. Comparative RFLP mapping of meadow and tall fescue., 1998, 97(1/2): 255-260.
[19] Huang L K, YAN H D, ZHAO X X, ZHANG X Q, WANG J, FRAZIER T, YIN G, HUANG X, YAN D F, ZANG W J, MA X, PENG Y, YAN Y H, LIU W. Identifying differentially expressed genes under heat stress and developing molecular markers in orchardgrass (L.) through transcriptome analysis., 2015, 15 (6): 1497.
[20] 李季, 黄琳凯, 金梦雅, 冯光燕, 赵欣欣, 聂刚, 潘玲, 唐露, 张新全. 鸭茅基因组Genomic-SSR标记开发. 分子植物育种, 2017, 15(10):1-11.
LI J, HUANG L K, JIN M Y, FENG G Y, ZHAO X X, NIE G, PANG L, TANG L, ZHANG X Q. Development and verification of orchardgrass Genomic-SSR., 2017, 15(10):1-11. (in Chinese)
[21] Vision T J, Brown D G, Shmoys D B, DURRETT R T, TANKSLKY S D. Selective mapping: a strategy for optimizing the construction of high-density linkage maps., 2000,155(1): 407.
[22] LIU D Y, MA C X, HONG W G, HUANG L, LIU M, ZENG H P, DENG D J, XIN H G, SONG J, XU C H, SUN X W, HOU X L, WANG X W, ZHENG H K. Construction and analysis of high-density linkage map using high-throughput sequencing data., 2014, 9(6):e98855.
[23] HUMPHREYS M W, YADAV R S, CAIRNS A J,TURNER L B, HUMPHREYS J, SKøT L. A changing climate for grassland research., 2006, 169: 9-26.
[24] 谢文刚, 刘文献, 张建全, 王彦荣. 牧草分子遗传连锁图谱及其应用. 草业科学, 2014, 31(6): 1147-1159.
XIE W G, LIU W X, Zhang J Q, WANG Y R. Molecular genetic linkage map and its application in forage crops., 2014, 31(6): 1147-1159. (in Chinese)
[25] JIANG Z Y, YU X X, YU Z, LIU Z H, HAO Z M, LI X L. Construction of an AFLP-based genetic linkage map of tetraploid hybrid wheatgrass., 2015, 35(4): 457-463.
[26] GUAN X L, HIRATA M, DING C, XU N, YUYAMA N,TAN L B, FU Y C, WANG J P, CAI H W. Genetic linkage map ofLam. constructed from a BC1 population derived from an interspecific hybridization,×L. ×., 2015, 60(3): 142-149.
[27] KING J, THOMAS A, JAMES C, KING I, ARMSTEAD I. A DArT marker genetic map of perennial ryegrass (L.) integrated with detailed comparative mapping information; comparison with existing DArT marker genetic maps of,, and., 2013, 14(1): 1-7.
[28] LU J, LU Y Y, LI J Q, ZHAN Q W, WANG M. SSR primer designing and construction of a genetic map of×., 2009, 31(2): 28-33.
[29] MORGANTE M, HANAFEY M, POWELL W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes., 2002, 30: 194-200.
[30] WANG Z, WEBER J L, ZHONG G, TANKSLEY S D. Survey of plant short tandem DNA repeats., 1994, 88(1): 1-6.
[31] 姜春芽, 廖娇, 徐小彪, 辜青青, 刘善军, 陈金印. 植物EST-SSR技术及其应用. 分子植物育种, 2009(1): 125-129.
JIANG C Y, LIAO J, XU X B, WEI Q Q, LIU S J, CHEN J Y. Plant EST-SSR technology and its application., 2009(1): 125-129. (in Chinese)
[32] WEN M F, WANG H Y, XIA Z Q, ZOU M L, LU C, WANG W Q. Developmenrt of EST-SSR and genomic-SSR markers to assess genetic diversity inL., 2010, 3(1): 42.
[33] 张亚东, 彭婵, 李振芳, 杨彦伶, 胡兴宜. 基因组SSR与EST-SSR标记在杨树不同种间的遗传差异. 东北林业大学学报, 2011, 39(12): 8-11.
ZHANG Y D, PENG C, Li Z F, YANG Y L, HU X Y. Genetic diversity of genomic-SSR and EST-SSR markers in interspecies of poplar., 2011, 39(12): 8-11. (in Chinese)
[34] 刘德新. 陆地棉遗传图谱加密与T_1区域纤维品质QTL精细定位及候选基因鉴定[D]. 重庆: 西南大学, 2015.
Liu D X. Maker density increase of genetic map and fine mapping and candidate identification of a major QTL controlling fiber quality traits at T1 region[D]. Chongqing: Southwest university. (in Chinese)
(责任编辑 李莉)
附表1 用于图谱构建的48对鸭茅SSR引物序列
Table S1 The 48 pairs of SSR primer sequences for mapping of orchardgrass

引物标号Primer上游引物Forward primer (5’-3’)下游引物Reverse primer(5’-3’) E45TGTCTTGTCAACAGCCGTGTGCTCCCCTAGGATTTCGTCG E107TTGCTTTTCACCCAGCCAATCTGGATCTCTCCTCTCCGGT E134TATTGTGCCCAGCGACTCTGCCCCTCTAGCCCTCTTCTCA E146GGCGCAGATCCTGTTACTGTCTGTATGCGCTCCTCTCCTG E168GTCCTTCCGAACCTTGGGAGCGGGGTAGACAGCTGAAAGG E192CCGAAACCTATCCGACGTGTGCTCCCCTAGGATTTCGTCG E281TGTACTGCCCTCGAGTCTGTCCCCTCTAGCCCTCTTCTCA E310CCGACACTTTCAGGACAGCAGTTCCAACCACGCAAACCAA E334ATGCTGTATCGCTGCCTGTTCGTCCTCTGTTCTGTTGTCCA E337GATCCATCTCTGTCACCGGCCTGTGCTTCCTGGTTTTGCC E347AGCCTCATCGCATTTCCACAAGTGTCCAGATGAACGGCAT E350TCGGTCCGAGGGAGTATTGTGCACCATCGAGGCATTCAAG E378AGAACAAGGCGACGCTAACATTAGGAGGCCGCAGAATTCC E393GAAGAGGGTGGGGTTGTGAGGTGCCAGTATCTCCGTGCC E435GGTTAACCGAAGCACATGGCACACACAATCTCCAGCCTGG E457CGTGCGAACCAAGCAGAAAACTCTTGGTTTTGCGGGGTTG E458CGTGGCTGCTACTGGTACATTGGTCAGCGAGGTACAGAGA E472TTCGGCCGTTTGATCACGTAATCGACGCCATCAAGAGCTT E488CGGAGGAGGATGACGACATGGGCCCAGCACCAAATCAAAT E498GCGTTACATCATGGTGCCACCGGGCGGCATGTAAAATACC E504CGCCATGGGTACTTCCTGTTCGTCCGATTCAGAAGAGCGA E527AGACAGAGGAAGCAGGGACAGTGCAAACACAAGACGGCAT E577TGAACTGTGCTGGACGACTCCTGTCACGCTCTGCTTACCA E612TCTGACACAAGGAATGCCGAGGTGGCTAATTGTGGACTCCA E644GGCTAAGACGACGTCTACGGAGAGAAGAGCAGAGCAGGGA E646TGGCGTTTGAAAGCAACACAATTTCTGGGATGAGCCTGCG E654CGTTGAAGTGCGATTGACCCCGTCGTCATTTCCTCCGTCA E711TTGGCGTCGAAGAGCTTCACAAACCCAATCCTCCCGCAAT E714CGTCAAGCCCTCATCCAACTTGGAAGAACCACGAGCAGTC E722GCTGCCAAGCTCAAACGAATACATCTCCAAGAGCACCACG E732GGGATTCCGACTCCGATTCCGTGCTCTCCAGGTTCATCCC G3GAATGGTCATTAGCATCCCTCAAGGAGCCAAAAATCTAAGTGGGGAGT G28AGAAGCGCCTCTTGATCATATCGCTCTTCCTAACCAAACCCTTCCC G30CGGAATTTATTATTTTAGGCCGCAGGAGCTGCTTGGAGAGCGAC G34AGCATCCTAGGCCGCTAATAAAACATGCAAATTAATGGTCCTTTTTGG G39GAGCTCACGATCATCCACGTCTACTCGTGGTGGCGAGAGCTAAT G40GCAGGAAACCACAGGAACAGTATTTGTTCAAGAATTTTCAGGTCAGCA G43GTGTGCAGAGTTCCTTCATTTCAGACTGTGCTTCTGAATTAATCATTCGT G47CCTCCATGTCCTCCTCCTTCTTAGGCAGAGTCGGTGCAGTATCT G48GATACTTCTCCCCAAATCCCAATCGTAGTCCACACCAGCGACCATAAC G60GCCGCTCTCTGGAGGATAAGACGCAGAGGAGAAGAGTAGAAGGAG G62GATGCCACTAAGGAGGATGAAGAGATACAAACTGAAGACACACCGCAA G72GCCCTAGTTTGAGATCACCATGACGTGCGATCCTGCAGAAAAACTAAC G76CCTTACCTCAGTGCTCCAACAGATCGACCAAGGAATCATACACACGTA G99TGAGGAGAGAGAGAGAGGAGTTGCTTCCTTCGTAGGGCTTTCCTCTT G96CGCGTATTCTCTCTTCTCTCTGCTCAGGGGTACTTCGGTCATTCTCTA G101TTTATCCCTGTTTGCCAGAGATTCCAACCGGTACCAAGTACTCGTGAT G112CAAAAGCCCTTCTATTCTACCCCATCCACTCTCCCTATGTTTCATTGC
附表2 第1连锁群120—150 cM区间内的标记位点
Table S2 Marker position from 120 cM to 150 cM on group 1 of updated genetic linkage map
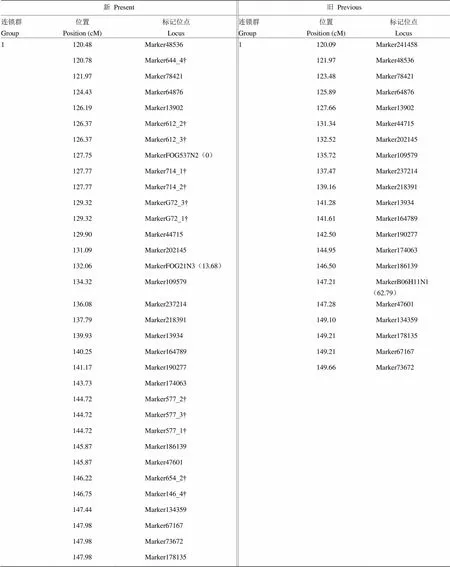
新Present旧Previous 连锁群Group位置Position (cM)标记位点Locus连锁群Group位置Position (cM)标记位点Locus 1120.48Marker485361120.09Marker241458 120.78Marker644_4†121.97Marker48536 121.97Marker78421123.48Marker78421 124.43Marker64876125.89Marker64876 126.19Marker13902127.66Marker13902 126.37Marker612_2†131.34Marker44715 126.37Marker612_3†132.52Marker202145 127.75MarkerFOG537N2(0)135.72Marker109579 127.77Marker714_1†137.47Marker237214 127.77Marker714_2†139.16Marker218391 129.32MarkerG72_3†141.28Marker13934 129.32MarkerG72_1†141.61Marker164789 129.90Marker44715142.50Marker190277 131.09Marker202145144.95Marker174063 132.06MarkerFOG21N3(13.68)146.50Marker186139 134.32Marker109579147.21MarkerB06H11N1(62.79) 136.08Marker237214147.28Marker47601 137.79Marker218391149.10Marker134359 139.93Marker13934149.21Marker178135 140.25Marker164789149.21Marker67167 141.17Marker190277149.66Marker73672 143.73Marker174063 144.72Marker577_2† 144.72Marker577_3† 144.72Marker577_1† 145.87Marker186139 145.87Marker47601 146.22Marker654_2† 146.75Marker146_4† 147.44Marker134359 147.98Marker67167 147.98Marker73672 147.98Marker178135
“†”表示新增的SSR标记;“()”内数字表示标记对应的旧(新)图谱所在位置
“†”Represent the new SSR marker and the number in the “()”represent the location of the previous (update) map corresponding to the mark

附图1 鸭茅各连锁群分子标记遗传图谱
Fig. S1 Distribution of SLAF and SSR markers on the seven linkage groups
Enhancement of the Genetic Linkage Map Density of Tetraploid Based on SSR Markers
TANG Lu, JIN MengYa, HUANG Linkai, ZHANG Xu, ZHAO Xinxin, ZHANG Xinquan
(Animal Science and Technology College, Sichuan Agricultural University, Chengdu 611130)
【Objective】In order to obtain a high density genetic map of tetraploid orchardgrass previously established, we used EST-SSR and genomics-SSR markers to enhance the density of the genetic map. These results will be beneficial and helpful to orchardgrass selection and QTLanalysis,especially QTL analysis of recessive genes.【Method】Based on the proposed test-hybridization strategy, an F1population of 214 individuals derived from the cross between two Chinese orchardgrass cultivars–Kaimo (tall height plant, more tillers, broad leaves and early-maturing) and 01436 (dwarf, less tillers, narrower leaves and late-maturing) was used for map construction. 574 pairs of EST-SSR markers and 150 pairs of Genomic-SSR markers were selected as the screening primers. Five of the 214 progenies were randomly selected and amplified together with their parents. Amplified fragments were separated on 8% denatured polyacrylamide gels. The primers which could amplify clear bands and the presence of separated polymorphic were used for population and parental DNA amplification. The amplified results were statistically analyzed according to the marker type. According to the presence or absence of bands (with band count 1, or count 0), the amplification products of the DNA were statistically analyzed. According to theχ2test, the marker which separation ratio was in accordance with 1﹕1 (Aaaa × aaaa or aaaa × Aaaa) and 3﹕1 (Aaaa × Aaaa) for genetic linkage map construction by using HighMap software.【Result】Finally, 31 pairs of EST-SSR primers and 17 pairs of Genomic-SSR primers were used for population and parental DNA amplification, The primer polymorphism percentage were 5.4%, 11.3% and 6.6%.A total of 169 clear bands were obtained, and 89 were used to construct the genetic linkage map of orchardgrass. There were 79 markers with Aaaa × aaaa or aaaa × Aaaa segregation types, 10 with Aaaa × Aaaa and the remaining 80 with distorted markers. A high-density linkage map of orchardgrass was constructed using 2,551 markers, which were distributed on seven linkage groups spanning 758.4 cM. The encrypted map including 4187 SNP markers, 84 SSR markers, the number of markers in the LGs from166 to709, with average 364. LG1 contains the largest maker with 709, while the LG7 was the least with 166. The sizes of the individual LGs ranged from 60.28 to 147.09 cM, with average inter-marker distances ranging 0.19—0.76 cM. The average distance between adjacent distance markers was reduced from 0.37 cM to 0.3 cM. Due to the change of marker density, the position of the markers distribution on each linkage group also changed greatly.【Conclusion】A high-density genetic linkage map of tetraploidwas reconstructed using 2 551 markers, which were distributed on seven linkage groups spanning 758.4 cM.The new map added a number of SSR markers, which total length increased by 42.63 cM and average distance between adjacent distance markers was reduced from 0.37 cM to 0.3 cM.
orchardgrass; ssr; genetic linkage map
2017-09-26;
2017-11-21
国家自然科学基金(NSFC 31372363)、国家现代牧草产业技术体系(CARS-34)
唐露,E-mail:15281737065@163.com。金梦雅,E-mail:anmnar@163.com。唐露和金梦雅为同等贡献作者。
张新全,E-mail:zhangxq@sicau.edu.cn
