黑土细菌及真菌群落对长期施肥响应的差异及其驱动因素
2018-03-26王慧颖徐明岗周宝库马想段英华
王慧颖,徐明岗,周宝库,马想,段英华
黑土细菌及真菌群落对长期施肥响应的差异及其驱动因素
王慧颖1,徐明岗1,周宝库2,马想1,段英华1
(1中国农业科学院农业资源与农业区划研究所/耕地培育技术国家工程实验室,北京 100081;2黑龙江省农业科学院土壤肥料研究所,哈尔滨 150086)
【目的】研究长期施肥对黑土细菌和真菌群落结构影响差异,探索黑土肥力对长期施用化肥和有机肥响应差异的生物学机制,为黑土的肥力培育和合理施肥提供科学理论依据。【方法】基于35年的长期定位施肥试验,采用定量PCR方法和Miseq高通量测序技术,分析长期不施肥(CK)、氮肥(N)、有机肥(M)和有机无机配施肥(MN)处理下,黑土细菌及真菌的数量、群落结构和多样性的差异。同时结合土壤理化性状,探究不同施肥条件下细菌和真菌群落变化的环境驱动因子。【结果】N处理对土壤细菌的数量没有显著影响,但使其群落多样性降低了13.2%—48.5%。N处理使真菌的数量增加了24倍,多样性降低了4.6%—80.3%。与N处理相比,MN处理使细菌数量和多样性分别增加了2倍和7.7%—46.6%,而真菌的数量虽降低了14.2%,但其多样性提高了62%—237%。单施氮肥增加了土壤细菌酸杆菌门()中的、及变形菌门()中的的相对丰度,并使土壤真菌中伞菌纲()的相对丰度增加了41倍。与N处理相比,MN处理下细菌的各主要类群丰度未发生显著变化,但M处理下土壤细菌中的、和丰度分别显著降低了26、97和81个百分点,、和的丰度分别显著增加了11倍、9倍和2倍。细菌群落结构在MN与N处理之间无显著差异,明显区别于CK和M处理,pH为主要驱动因素,其阈值为6.07;真菌群落结构在CK、M和MN处理下相似,显著区别于N处理,两组处理之间差异由速效钾含量(125.5 mg·kg-1)驱动。另外,有机质含量对于细菌和真菌群落均是重要的驱动因素,但调控细菌群落结构的阈值为28.4 g·kg-1,而驱动真菌群落结构的阈值为30.8 g·kg-1。【结论】黑土细菌对有机肥的响应较强,而真菌对化肥更为敏感。长期施用化肥会刺激土壤中嗜酸细菌和真菌的生长,而有机无机肥配施可提高土壤微生物群落多样性,刺激有益菌的生长。土壤pH和有效钾含量分别是调控细菌和真菌群落结构的重要影响因素,在黑土肥力培育中应引起充分的重视。
细菌;真菌;长期施肥;mesiq测序;qPCR;黑土
0 引言
【研究意义】东北黑土区是中国商品粮生产基地,其黑土也是全球少有的几种肥沃土壤之一。但近些年黑土退化严重、土壤肥力下降、土壤酸化及粮食产量降低等问题受到了广泛关注。曾有学者认为黑土退化主要由于黑土由自然生态系统向农田生态系统转化过程中生态结构发生变化,更重要的是人为非科学活动所致[1]。向土壤中人为大量输入外源物质,尤其肥料,被认为是导致农田生态系统稳定性及土壤肥力发生变化的关键因素[1-2]。微生物通过参与有机质的降解、腐殖质的形成、养分的转化以及碳氮磷等的元素循环从而改善土壤肥力与生产力[3-4]。同时作为土壤中重要的生命有机体,微生物生物学特性和群落结构与土壤质量有密切关系,它们对生存环境的改变极其敏感,微生物数量、多样性和群落结构等均能作为判断土壤健康状况的重要指标[5]。另外,微生物群落结构的演变是一个漫长的过程,很多驱动微生物群落响应的过程会随施肥年限增强或减弱[6]。因此,研究长期定位施肥下的微生物群落变化及其驱动因素对提升黑土肥力具有重要意义。【前人研究进展】细菌和真菌是土壤中主要的两大微生物类群[7]。近些年关于长期施肥对土壤微生物群落变化的影响及其驱动因素的研究大都集中在细菌[8-10],对于真菌及其与细菌之间的关系在农田生态系统中的重要作用,尚有待进一步探索。真菌细胞中的碳氮比(C/N)远高于细菌细胞,需要从土壤环境中摄取更多有机物质作为碳源的营养来源,因此较细菌群落更容易影响土壤肥力[11-12]。另外,土壤的有机质分解和养分转化等重要过程主要由细菌和真菌共同完成,如土壤中大多数复杂、难分解的有机质的分解由真菌完成,而简单、易被分解的有机质由细菌分解[13-14]。同时,细菌与真菌在养分利用中还存在协作和竞争等关系。在养分匮乏的土壤环境中,真菌的菌丝有利于提高并均衡土壤中的养分,进而促进细菌的生长代谢[15-17]。近年来,ZHOU等[7,18]、秦杰等[19]等应用分子生物学手段研究了化肥对黑土区细菌和真菌群落的影响,且取得了突破性进展,其结果发现长期施用化肥导致黑土细菌和真菌群落发生了显著变化,氮肥导致黑土从“细菌型”向“真菌型”转变,且高量化肥对细菌和真菌的影响比低量化肥的影响更显著。然而,之前的研究主要分析了细菌和真菌群落对不同化肥施用量的响应,但其对有机肥和混施肥的响应及其主要驱动因素,尚缺乏深入研究。【本研究切入点】长期施用化肥可降低土壤pH进而显著影响细菌与真菌群落[17-18],而有机肥料的施用不仅向土壤中接种了大量的外源微生物直接影响土壤微生物[20],且为微生物生长提供碳源和能源,其养分的缓慢释放还可为微生物提供更稳定的栖息环境[8-10],进而间接影响微生物群落。然而,长期化肥和有机肥处理下,黑土细菌和真菌群落的响应差异,尤其是不同施肥影响了哪些关键菌群,进而调控了土壤养分循环,亟需进行深入分析。【拟解决的关键问题】本文基于黑土上连续35年的长期定位施肥试验,采用定量PCR和Miseq高通量测序方法,探究细菌和真菌的数量、群落结构和多样性对长期施用不同肥料的响应差异,并结合土壤理化性状揭示其驱动因素,以期阐明不同施肥管理措施影响黑土肥力的生物学机制,为黑土的肥力培育和合理施肥提供科学理论依据。
1 材料与方法
1.1 试验时间和地点
长期定位施肥试验设于农业部哈尔滨黑土生态环境重点野外科学观测试验区内(N 45°40′,E126°35′),该区属于典型的季风性气候,年平均降雨量为575 mm,年平均蒸发量为1 315 mm,年平均气温为3.0℃。未开展试验前,土壤pH和有机质(SOM)的初始值分别为7.45和27.0 g·kg-1;全氮(TN)、全磷(TP)和全钾(TK)的初始值分别为1.48 g·kg-1、1.07 g·kg-1和25.3 g·kg-1;碱解氮(AN)、速效磷(AP)和速效钾(AK)分别为149.2 mg·kg-1、51 mg·kg-1和210 mg·kg-1。试验设于1979年,始于1980年并按小麦-大豆-玉米进行轮作,且为一年一熟制。试验共设8个处理,3次重复,每个小区面积36 m2(9 m×4 m),各重复随机区组排列。本试验选取其中4种典型处理进行研究:不施肥(CK)、单施化学氮肥(N)、单施有机肥(M)、化学氮肥和有机肥混合施用(MN),其中化学氮肥为尿素,其施用量在大豆茬为75 kg·hm-2,在小麦和玉米茬的施用量均为150 kg·hm-2;有机肥为纯马粪(有机质28.2%,氮素0.473%,磷0.458%,钾0.63%),每轮作周期在玉米根茬施用一次,其用量为18 600 kg·hm-2。
1.2 试验材料
土壤样品为2014年10月所采集的耕层土壤(0—20 cm),每个处理的每个重复随机采9个点,混匀成1个样品,且每个重复采集2个土壤样品。剔除土样中的石粒和植物残根等杂物,装入无菌封口袋后暂时贮存在低温冰盒中,并迅速带回实验室;过2 mm筛后的土样分成两部分,一部分风干处理,用于土壤理化性状分析;另一部分保存在-80℃冰箱,用于土壤DNA的提取。
1.3 试验方法
1.3.1 土壤理化性状分析 土壤pH采用1﹕2.5的水土比、复合电极测定;有机质含量采用重铬酸钾-硫酸外加热方法测定;全氮采用半微量凯氏定氮法;全磷全钾采用硫酸-高氯酸消煮后分别用钼锑抗比色法和火焰光度计法测定;速效磷采用NaHCO3浸提-钼锑抗比色法;速效钾采用HF-HClO4消煮-火焰光度计法;铵态氮及硝态氮用KCl浸提后利用流动分析仪测定;碱解氮采用扩散法测定,即用碱液处理样品,硼酸吸收后以标准酸滴定。以上测定方法均参考《土壤农业化学分析方法》[21]。
1.3.2 土壤DNA的提取及qPCR,Meiseque分析 用Powersoil DNA Isolation Kit(MoBio Labories,Carlsbad,CA,USA)试剂盒从0.25 g土壤样品中提取60 μl DNA。避免提取过程中的误差,利用DNeasy Tissue kit(Qiagen,Valencia,CA,USA)将得到的DNA进行纯化。根据260/280-nm和260/230-nm吸收比例,用NanoDrop ND-2000分光光度计(NanoDrop,ND2000,Thermo Scientific,111 Wilmington,DE)测定土壤DNA的质量。以341F(5′-CCTACGGGNG GCWGCAG-3′)和 805R(5′- GACTACHVGGGTATC TAATCC-3′)为DNA引物扩增代表细菌的16S rRNA序列[22];以为ITS2(5′-GCTGCGTTCTTCATCGATGC -3′)和ITS1(5′-CTTGGTCATTTAGAGGAAGTAA -3′)DNA引物扩增定代表真菌的ITS rRNA序列[23-24]。利用ABI Real-Time 7500 System(Applied Biosystems,Waltham,MA,USA)及SYBR Green(FasteFire Qpcr Premix,TIANGENBIOTECH,China)进行qPCR的克隆扩增反应,反应条件见表1。利用Minipre Kit(Qiagen,Germantown,MD,USA)得到所需的质粒DNA,并以此拷贝数及2>0.99为标准进行qPCR反应,且每个被测DNA样品设置3次重复。
采用Illumina Miseque Platform System的Miseque 高通量测序方法测定代表真菌和细菌的ITS和16S序列。对于得到的高通量序列的结果,首先去除测定序列中的杂质,然后以10 bp的barcodes及引物序列为标准对序列进行修剪。在分析过程中,去除长度小于200 bp和包含未被处理的核酸序列;随后,真菌和细菌的每条序列分别在以SRX766215和SRX1032809为检索号的NCBI数据库中进行匹对。最后,分别得到24个样品的8 703—9 820条不等的真菌序列和10 405—13 642条不等的细菌序列;将每个样品的真菌和细菌序列统一纯化为8 703和10 405条。以97%的序列相似度为标准,将所有序列组成不同的可操作单元(OTUs);最后得到代表真菌序列的837个OTUs,代表细菌序列的5 075个OTUs。以85%的置信水平为标准,利用Ribosomal Database Project(RDP)平台将每个代表序列的OTU进行物种匹对,得到每个OTU所代表的物种。
表1 定量PCR反应条件
1.3.3 数据分析 将qPCR反应得到的ITS rRNA及16S rRNA基因拷贝数取log后的值作为细菌和真菌的相对数量,二者拷贝数的比例作为真菌和细菌的数量比(F/B)。细菌和真菌的α-多样性通过计算Chao1,香农(Shannon)和辛普森(Inv_Simpson)指数获得。原始数据首先经EXCEL2007软件处理,随后采用SPSS16.0软件(SPSS,Chicago,IL,USA)的描述统计(Descripitive Statistics)将原始数据进行标准化;利用单因素方差分析(One-way ANOVA)中的Duncan方法对不同施肥处理的土壤理化性状、微生物数量和多样性、每种菌的丰度进行方差分析和显著性检验;采用R语音软件的2.15.3版本对不同施肥处理间的OTUs及土壤因子进行对应分析(CA)和多元回归树(MRT)分析。以上所有分析的显著性检验标准为<0.05。
2 结果
2.1 长期不同施肥对土壤理化性状及作物产量的影响
所有施肥处理均显著提高了土壤中除有效钾外的养分含量(表2)。在施肥处理中,所有养分含量均为MN处理最高。与N处理相比,土壤的SOM、TN、AN、AP和AK含量在MN处理中分别显著增加了17%、17%、19%、263%和16%;与M处理相比,土壤的SOM、TN、AN、AP和AK含量在MN处理下分别显著增加了15%、21%、41%、195%和12%。土壤pH值在施用化肥的处理(MN和N)下较CK处理显著降低了1.2—1.4个单位,而在只施用有机肥处理下未显著变化。一轮作周期内作物的年均产量在MN和N处理下最高,M处理其次,在不施肥处理最低。
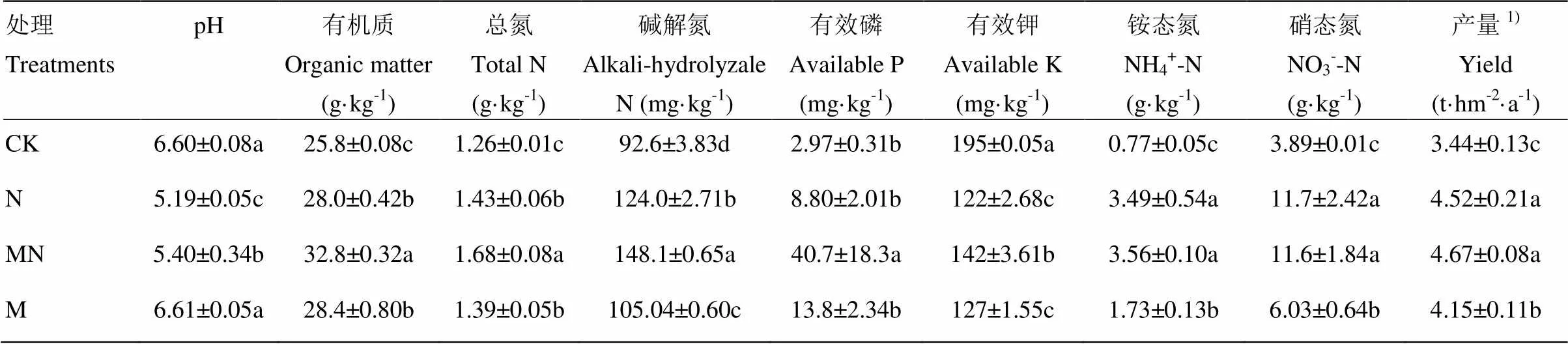
表2 长期不同施肥下黑土的理化性状及产量
1)产量代表轮作一周期内三种作物产量的均值,即当年大豆产量与之前两年作物(玉米和小麦)的均值;同列数据后不同小写字母表示处理间土壤的理化性状及作物产量差异达到显著水平(<0.05)
1)Yield indicated the average value of three typical crop yields during a latest rotation system; Values followed by lowercases in the same column mean significant differences in soil properties and crop yield between treatments (<0.05)
2.2 长期不同施肥下细菌和真菌的数量及多样性
由表3可见,细菌数量和真菌数量均在M处理下最高。与CK、N和MN处理相比,M处理的细菌数量分别显著增加了2.74倍、4.20倍和0.91倍,真菌数量分别显著增加了50倍、1.04倍和1.38倍(<0.05)。与CK处理相比,N处理的细菌数量无显著差异,但真菌数量显著增加了24倍(<0.05)。细菌数量在MN处理下较N处理下显著增加了1.72倍,而真菌数量在MN与N之间没有显著差异。真菌与细菌相对数量比(F/B)在N处理最高,其次为MN和M处理,CK处理最低。
香农指数(Shannon)、辛普森指数(Inv_Simpson)和Chao1指数均是表征微生物α-多样性的指标,其值越高表示微生物多样性越高。细菌的多样性指数在4个处理中差异显著,香农指数、辛普森指数和Chao1指数的变化趋势为:M≥CK>MN>N,其中混施肥MN处理比N处理的α-多样性提高了7.7%—46.6%。真菌的α-多样性指数在CK、MN和M处理中无显著差异,且较N处理的α-多样性指数显著提高了28.9%—237.3%(<0.05)。
2.3 长期不同施肥下细菌和真菌的群落结构
图1是分别以组成24个土样细菌和真菌的5 076和837个OTUs数据为基础得出的对应分析(CA)结果。CA1轴可解释65.8%细菌和61.5%真菌群落结构的变化,且其群落结构和组成在不同施肥处理间差异较大。黑土细菌和真菌群落结构均在CK处理与M处理相似,并在CA1轴上与施N处理分开。细菌和真菌群落结构对长期混施肥MN处理的响应却不一致,细菌群落结构在MN处理下与N处理下相似,共同位于CA1轴的右半轴;而MN处理下的真菌群落处于CA1轴中间,在CA2轴明显与其他3个处理分开。
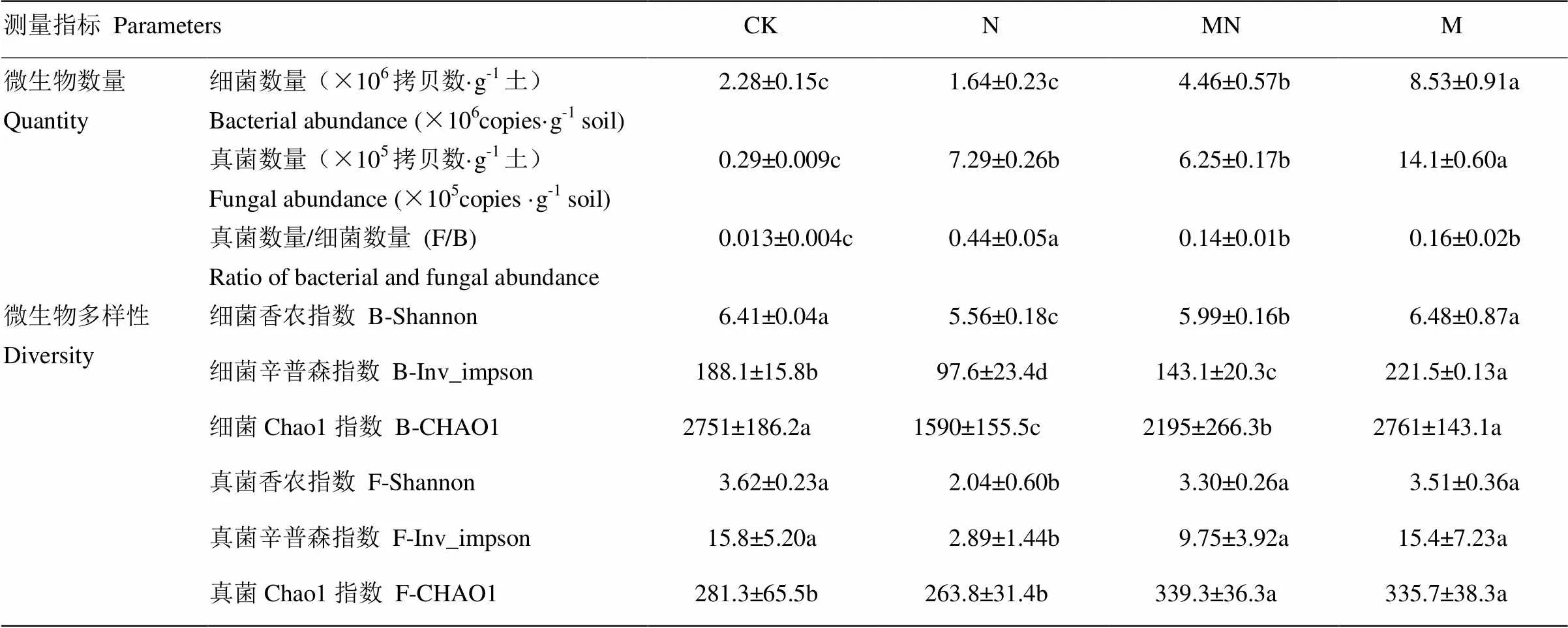
表3 长期不同施肥下黑土的细菌和真菌数量及其多样性
同行数据后不同小写字母表示处理间土壤的微生物指标差异达到显著水平(<0.05)
Values followed by lowercases in the same rows mean significant differences between treatments (<0.05)

图1 长期不同施肥对土壤细菌群落及真菌群落结构影响的对应分析(CA)
长期不同施肥处理下,在纲水平相对丰度大于2%的细菌有16个,纲水平相对丰度大于3%的真菌有9个,图2分别为这些细菌的相对丰度和真菌的相对丰度。与CK处理相比,M处理的各细菌相对丰度未发生显著变化;而MN和N处理的土壤酸杆菌()中的和及α-变形菌纲()均显著增加(<0.05),其中MN处理使得它们分别增加了36倍、4.3倍和0.35倍,N处理使得它们分别增加了11倍、3.1倍和0.52倍。同时,MN处理的_、和浮霉菌纲()的丰度显著比CK处理下降低了75%、75%和28%,但显著比N处理下提高了262%、147%和27%。
在9种主要纲水平的真菌中,伞菌纲()的相对丰度在各处理中的差异最显著,其变化趋势为:N>MN>M>CK,且在N处理的真菌群落中的相对丰度高达70%,比在CK处理下提高了41倍;粪壳菌()和毛霉菌()在M和CK处理下的丰度最高,其次为MN,N处理中最低。

图2 长期不同施肥下细菌和真菌的主要群落组成
2.4 土壤养分含量与细菌及真菌群落结构的关系
不同处理中,MN和N处理的作物产量与土壤养分含量均处于较高水平(表2),且二者的细菌和真菌群落结构均与CK处理有显著差异(图1,图2)。因此,表4分析了与CK处理相比,MN和N处理具有相同显著变化趋势的纲水平细菌和真菌的丰度与土壤理化性状的相关性。表中所有细菌和真菌的相对丰度均与土壤pH呈极显著相关关系(<0.01),其中、、和与土壤pH呈极显著负相关;、、和与pH呈极显著正相关。同时,我们发现土壤养分(除AK外)含量均与丰度呈显著正相关(<0.05);SOM、AN、NH4+和NO3-含量与中的、呈显著正相关,与、呈显著负相关(<0.05)。真菌中丰度与NH4+含量呈显著正相关,而和与AN、NH4+和NO3-含量呈显著负相关(<0.05)。
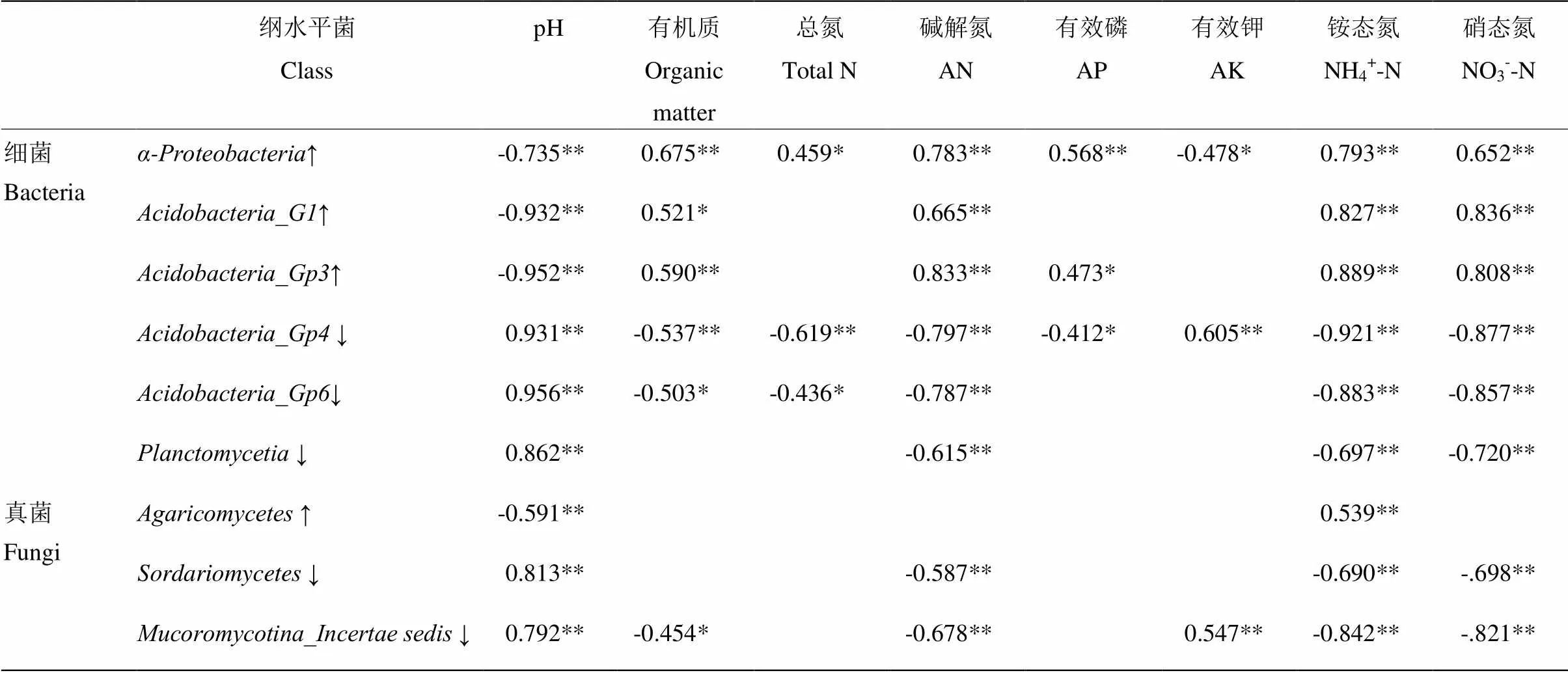
表4 长期施肥下显著变化的细菌或真菌丰度与土壤理化性状之间的相关分析
纲水平菌:与CK相比,MN和N处理下具有相同显著变化趋势的细菌和真菌。其中“↑”或“↓”代表此物种的丰度在MN和N处理下均显著提高或降低。“**”和“*”代表显著性水平分别为<0.01和<0.05,且显著性水平≥0.05的相关系数未列入表中
Class: Compared with CK, bacterial or fungal abundance commonly and significantly changed under MN and N treatments. “↑” or “↓” indicated this species abundance commonly and significantly enhanced or decreased. “**” and “*” indicated difference were significant at the levels of<0.01 and<0.05, respectively. Correlation coefficient at the level of≥0.05 was not showed in form
根据“1SE”准则,选择最小交叉验证相对误差CVRE值[25]将微生物群落进行多元回归分析,以明确驱动黑土细菌和真菌群落的主要因子(图3)。4个处理的细菌和真菌群落均被分成了3组:CK与M、N和MN。土壤因子对细菌和真菌群落结构变化的解释率(2)分别为76.8%和67.3%。细菌群落的主要驱动因子为土壤pH值,其阈值为6.07;在pH低于6.07的土壤中,细菌群落结构进一步受SOM含量影响,其阈值为28.4 g·kg-1。真菌群落的主要驱动因子为土壤AK含量,其阈值为125.5 mg·kg-1;在AK含量高于125. 5 mg·kg-1的土壤中,真菌群落结构也受到SOM含量影响,其阈值为30.8 g·kg-1。
3 讨论
土壤微生物是农田生态系统的重要组成部分,施肥和肥料类型与土壤微生物的数量、组成及多样性的差异紧密相关[26-27]。本研究结果表明,长期施用氮肥未显著改变土壤细菌数量,但增加了真菌数量,因此土壤微生物向F/B值较高的“真菌型”转变(表3),这与前人提出的相比细菌,真菌更适宜生长在施用化肥的环境中的结果一致[18,28]。其原因可能主要有:一是长期施用氮肥导致土壤养分较为单一,不能满足部分细菌生长代谢所需,然而,以菌丝生长方式繁殖的真菌能够利用广泛分布的菌丝从作物根茬或其他养分源中获得大量且均衡的养分供自身代谢[17],因此其生长速度相对较高;二是与适宜生长在pH为7左右且与对土壤pH变化敏感的细菌相比,真菌对酸化土壤环境的适应能力更强[29],因此单施化肥更利于真菌的生长。MN处理的细菌数量和多样性均显著高于N处理,说明化肥配施有机肥可促进细菌的生长与繁殖[30-31]。可见细菌与真菌的生长与肥料类型密切相关,调控肥料类型与比例是协调不同类型微生物生长和提高土壤生物肥力的有效途径。
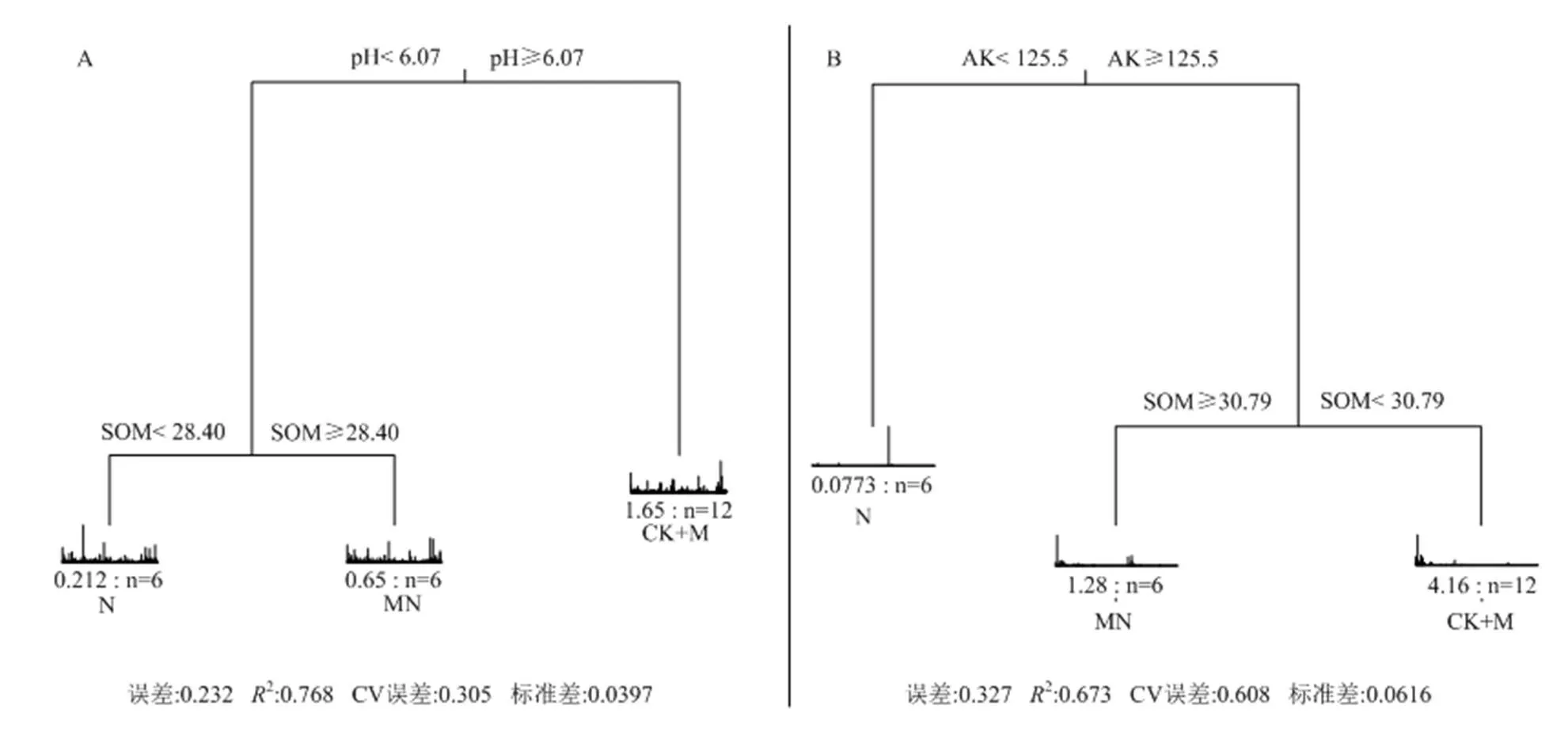
图3 驱动细菌群落(A)和真菌群落(B)结构变化的土壤因子的多元回归树(MRT)分析
与不施肥相比,单施化肥尽管显著提高了真菌数量且未显著影响细菌数量,但却显著降低了细菌和真菌的多样性(表3)。这是由于长期施用氮肥导致pH较低和养分不均衡刺激了嗜酸和嗜氮微生物的生长[28,32]。同时,生物间的竞争性导致其他微生物的生长变慢,甚至消失,因此微生物多样性降低。此外,真菌在N处理的数量大于CK处理,而细菌的数量在两个处理中相近,说明嗜氮微生物可能大量存在于真菌群落中,或真菌群落中的嗜氮微生物繁殖速率高于细菌群落中的嗜氮微生物繁殖速率。与单施化肥(N)相比,施用有机肥(MN和M处理)显著提高了细菌和真菌的多样性(表3)。这可能是由于有机肥一方面改善了由于N处理引起的酸化和养分不均衡的土壤环境[33-34],有效减缓了嗜氮和嗜酸微生物对其他微生物的抑制作用;另一方面,有机肥向土壤中输入了大量的外源微生物[20,35],从而提高了微生物多样性。
由于微生物会随生态位点及作用底物的差异而发生变化,故不同施肥处理所引起的土壤环境变化显著影响了微生物群落的组成[29-30]。与各处理细菌的丰度变化相比,真菌的变化程度更大(图2),尤其是真菌担子菌()中的,其在施肥处理较不施肥处理提高了41倍。多数属于丛植菌根真菌,能为作物提供大量养分,促进作物生长[36]。且多为腐生菌,能分泌过氧化物酶从而分解和利用土壤中木质素、植株残留物等有机质[37],与CK处理的低有机质环境相比,有机质水平较高的施肥处理为提供了生长代谢底物,因此显著提高了的丰度(表4,图2)。其中化肥处理显著提高了的丰度,这与Wang等[38]的结果一致。我们发现化肥处理下相对丰度的提高程度比有机肥处理的提高程度更大,但Wang[38]的结果却表明化肥对的影响并不显著。这可能因为的丰度与土壤pH呈负相关,与NH4+呈正相关[18](<0.01,表4),所以长期化肥处理下,Wang等[38]所研究的pH只降低了0.09个单位,而NH4+降低了1 mg·kg-1的潮土环境对的影响不明显;而我们所研究的pH显著降低了1.41个单位,且NH4+提高了2.6 mg·kg-1的黑土环境更适合的生长。我们推测长期化肥处理的黑土提高了对养分和能源的竞争力,导致其他微生物生长缓慢甚至消失,因此真菌多样性在N处理下显著降低,但真菌数量却因的大量繁殖而显著增加。
在细菌群落中的比例高达20%,是土壤中主要的细菌菌群之一,对土壤的养分循环及微生态环境的平衡起着重要作用。JANSSEN等对美国87个土样的群落结构进行研究时发现,、、及为的主要类群[38-39]。本研究发现这4种酸杆菌纲不仅为的主要类群,而且均对长期施肥有明显响应(图2)。和在含化肥处理(MN和N)的土壤中显著高于CK和M处理,而和的变化趋势则相反。众多研究表明低pH的土壤环境更适合生长[39-41],本研究结果显示和与土壤pH呈极显著负相关(<0.01,表4),说明Gp1和Gp3为嗜酸菌,适宜生长在pH较低的土壤中。然而,和与土壤pH呈极显著正相关(<0.01,表4),说明它们可能更适宜生长在偏中性的土壤中。我们的研究结果中,长期MN和N处理显著提高了丰度,说明施用氮肥在黑土中可提高的丰度,这与Fierer等[42]的研究结果一致。同时,丰度与养分呈极显著正相关关系(<0.01,表4),这与前人认为变形菌()为富营养菌[43]的结果一致,但我们的研究未发现、这两种对环境改变更敏感的变形菌在不同施肥处理中发生显著变化,对此值得进一步深入的研究。
大量研究结果表明,土壤pH为陆地生态系统中影响细菌和真菌群落变化的关键因子[31,43]。本研究证明了pH不仅是影响黑土细菌群落的首要因子,且进一步提出了引起细菌群落变化的关键土壤pH阈值为6.07(图3-A)。长期施用化肥的N和MN处理下,土壤pH降至5.2—5.4,其细菌群落结构显著区别于pH为6.6的CK和M处理(表2,图3-A)。然而,与以往报道不同的是,本试验中驱动真菌群落变化的关键因子为有效钾(AK),其阈值为125.5 mg·kg-1(图3)。这可能是由于本研究中土壤pH变化范围较小(5.2—6.6),真菌群落对此范围内变化的pH并不敏感[31]。而且,在钾含量较低的N处理下(表2),作物与真菌竞争土壤中的AK,因此较低的AK含量成为N处理下真菌群落的限制因素。但是,同样处于较低AK水平的M处理中的真菌群落却未发生显著变化。我们推测其原因可能是:与N处理相比,一方面M处理利于真菌从有机肥中摄取更多的钾[34,44],另一方面M处理的作物产量比N处理低,作物与真菌间对AK的竞争变弱。
土壤有机质可同时调控黑土中的真菌和细菌群落结构。作为微生物的能量来源和代谢底物,土壤有机质的含量及其成分显著影响着微生物的群落组成[8,43]。调控黑土细菌和真菌群落结构的SOM阈值分别为28.4 g·kg-1和30.8 g·kg-1,这可能由于不同处理下土壤有机质组分的活性不同[45],且细菌和真菌群落对有机质的分解利用能力存在差异所致[15,46]。对于真菌来说,作物产量较高的MN处理土壤有机质含量较高,且大量来源于作物根茬,其成分多为复杂且难被分解的木质素等,更易被-策略的贫营养型真菌群落分解利用[15],故MN处理中的真菌群落显著区别于CK和M;对于细菌来说,有机肥中除了复杂有机物外,还含有大量的半纤维素和蛋白质等简单有机物,它们更易被-策略的富营养型细菌群落分解利用[15],故MN处理的细菌群落结构显著区别于N处理的细菌群落。因此,在不同有机质水平下,明确土壤中细菌和真菌的群落构成和主要功能菌的变化,将是下一步的研究重点。
4 结论
4.1 不同施肥方式通过影响土壤的理化性状,进而影响土壤细菌和真菌的数量及群落构成。长期施用化肥不会影响黑土中细菌的数量,但是降低了其多样性,其中主要是促进了、和的生长,而抑制了、和的生长。长期施用化肥可提高土壤真菌的数量却降低了其多样性,促进的生长。对于化肥配施有机肥,可显著提高土壤细菌的数量,并提高细菌和真菌的多样性。施用化肥可刺激土壤中真菌的生长,而有机肥对细菌的作用更加明显。
4.2 驱动黑土细菌群落结构变化的首要因素是土壤pH,其阈值为6.07,这也是施用化肥土壤(N和MN的pH分别为5.2和5.4)的群落结构与不施肥或仅施用有机肥的土壤(pH均为6.6)的群落结构差异较大的原因。土壤有效钾含量对于调节土壤真菌群落结构起着重要作用。土壤有机质含量对于细菌和真菌群落均起到重要的调节作用,但是其调控阈值不同。
[1] 李海波, 韩晓增, 王风. 长期施肥条件下土壤碳氮循环过程研究进展. 土壤通报, 2007, 38(2): 384-388.
LI H B, HAN X Z, WANG F. Review of soil carbon and nitrogen cycling under Long-term fertilization.2007, 38(2): 384-388.
[2] 隋跃宇, 张兴义, 焦晓光, 王其存,赵军. 长期不同施肥制度对农田黑土有机质和氮素的影响. 水土保持学报, 2005, 19(6): 190-192.
SUI Y Y, ZHANG X Y, JIAO X G, WANG Q C, ZHAO J. Effect of long-term different fertilizer applications on organic matter and nitrogen of black farmland.2005, 19(6): 190-192. (in Chinese)
[3] 魏丹, 杨谦, 迟凤琴. 东北黑土区土壤资源现状与存在问题. 黑龙江农业科学, 2006(6): 69-72.
WEI D, YANG Q, CHI F Q. The soil resource conditions and the problems in northeast black soil regions.2006(6): 69-72. (in Chinese)
[4] DENEF K, ROOBROECK D, WADU MWadu, M C W M, LOOTENS P, BOECKX. Microbial community composition and rhizodeposit-carbon assimilation in differently managed temperate grassland soils.2009, 41(1): 144-153.
[5] DEGENS B P, SCHIPPER L A, SPARLING G P, VOJVODICVUKOVIC M. Decreases in organic C reserves in soils can reduce the catabolic diversity of soil microbial communities., 2000, 32(2): 189-196.
[6] PENG C J, LAI S S, LUO X S, LU J W, HUANG Q Y, CHEN W L. Effects of long term rice straw application on the microbial communities of rapeseed rhizosphere in a paddy-upland rotation system., 2016, 557: 231-239.
[7] ZHOU J, GUAN D, ZHOU B,ZHAO B, MA M, QIN J. Influence of 34-years of fertilization on bacterial communities in an intensively cultivated black soil in northeast China., 2015, 90: 42-51.
[8] XUN W B, ZHAO J, XUE C, ZHANG G, RAN W, WANG B. Significant alteration of soil bacterial communities and organic carbon decomposition by different long-term fertilization management conditions of extremely low-productivity arable soil in South China., 2016, 18(6): 1907-1917.
[9] 魏巍, 许艳丽, 朱琳, 韩晓增. 长期施肥对黑土农田土壤微生物群落的影响. 土壤学报, 2013, 50(2): 372-380.
WEI W, XU Y L, ZHU L, HAN X Z. Effect of long-term fertilization on soil microbial communities in farmland of black soil.2013, 50(2): 372-380. (in Chinese)
[10] 马琳, 孙本华, 孙瑞, 高明霞, 杨学云. 长期不同施肥对土细菌群落多样性的影响. 西北农业学报, 2015, 24(6): 162-170.
MA L, SUN B H, SUN R, GAO M X, YANG X Y. Effect of long-term different fertilization on bacterial community diversity of lou soil., 2015, 24(6): 162-170. (in Chinese)
[11] GRANDY A S, STRICKLAND M S, LAUBER C L, BRADFORD M A, FIERER N. The influence of microbial communities, management, and soil texture on soil organic matter chemistry., 2009, 150(3/4): 278-286.
[12] HU S, FIRESTONE M K, CHAPIN F S. Soil microbial feedbacks to atmospheric CO2enrichment., 1999, 14(11): 433.
[13] MG V D H, BARDETT R D, STRAALEN N M. The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems., 2008, 11(3): 296-310.
[14] 陈永亮, 陈保冬, 刘蕾, 胡亚军, 徐天乐, 张莘. 丛枝菌根真菌在土壤氮素循环中的作用. 生态学报, 2014, 34(17): 4807-4815.
CHEN Y L, CHEN B D, Liu L, HU Y J, XU T L, ZHANG S. The role of arbuscular mycorrhizal fungi in soil nitrogen cycling., 2014, 34(17): 4807-4815. (in Chinese)
[15] STRICKLAND M S, ROUSK J. Considering fungal: bacterial dominance in soils - methods, controls, and ecosystem implications., 2010, 42(9): 1385-1395.
[16] SONNEMANN I, WOLTTERS V. The microfood web of grassland soils responds to a moderate increase in atmospheric CO2., 2005, 11(7): 1148-1155.
[17] VESTERGARD M, HENRY F, RANGEL-CASTRO J I, MICHELSEN A, PROSSER J I. Rhizosphere bacterial community composition responds to arbuscular mycorrhiza, but not to reductions in microbial activity induced by foliar cutting., 2008, 64(1): 78-89.
[18] ZHOU J, JIANG X, ZHOU B K, ZHAO B S, MA M CH, GUAN D W, LI J, CHEN S F, CAO M F, SHEN D L, QIN J. Thirty four years of nitrogen fertilization decreases fungal diversity and alters fungal community composition in black soil in northeast China., 2016, 95: 135-143.
[19] 秦杰, 姜昕, 周晶, 马鸣超, 关大伟, 周宝库, 赵百锁, 杜秉海, 李俊. 长期不同施肥黑土细菌和古菌群落结构及主效影响因子分析. 植物营养与肥料学报, 2015, 21(6): 1590-1598.
QIN J, JIANG X, ZHOU J, MA M C, GUAN D W, ZHOU B K, ZHAO B S, DU B H, LI J. Characteristics and driving factors of soil bacterial and archaeal communities under long-term fertilization regimes in black soil., 2015, 21(6): 1590-1598. (in Chinese)
[20] 于淑玲. 腐生真菌在有机质分解过程中的作用研究进展. 河北师范大学学报(自然科学版), 2003, 27(5): 519-522.
YU S L. A study of function that rot funguses have in the decomposition of organic matter.2003, 27(5): 519-522. (in Chinese)
[21] 鲁如坤. 土壤农业化学分析方法. 北京: 中国农业科技出版社, 2000.
LU R K.. Beijing: China Agricultural Science and Technology Press, 2000. (in Chinese)
[22] HERLEMANN D P, LABRENZ M, JÜRGENS K, BERTILSSON S, WANIEK J J, ANDERSSON A F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea., 2011, 5(10): 1571.
[23] KHOR W C, ROUME H, COMA M, HAN V, RABAEY K. Acetate accumulation enhances mixed culture fermentation of biomass to lactic acid., 2016, 100(19): 8337-8348.
[24] KORMAS K A, PACHIADAKI M G, KARAYANNI H, LEADBETTER E R, BERNHARD J M, EDCOMB V P. Inter- comparison of the potentially active prokaryotic communities in the halocline sediments of Mediterranean deep-sea hypersaline basins., 2015, 19(5): 949-960.
[25] BECK J J, LIGON J M. Polymerase chain reaction assays for the detection of Stagonospora nodorum and Septoria tritici in wheat., 1995, 85(3): 319-324.
[26] Romón P, ZHOU X, ITURRONDOBEIRIA J C, WINGFIELD M J, GOLDARAZENA A. Ophiostoma species (Ascomycetes: Ophiostomatales) associated with bark beetles (Coleoptera: Scolytinae) colonizing Pinus radiata in northern Spain., 2007, 53(6): 756-767.
[27] De'Ath G. De'Ath G. Multivariate regression trees: a new technique for modeling species-environment relationships., 2001, 83(4): 1105-1117.
[28] GEISSELER D, SCOW K M. Long-term effects of mineral fertilizers on soil microorganisms-A review., 2014, 75: 54-63.
[29] SUN L, XUN W B, HUANG T, ZHANG G, GAO J, RAN W. Alteration of the soil bacterial community during parent material maturation driven by different fertilization treatments., 2016, 96: 207-215.
[30] LING N, ZHU CH, XUE CH, DUAN Y H, CHANG P, GUO S W, SHN Q R. Insight into how organic amendments can shape the soil microbiome in long-term field experiments as revealed by network analysis., 2016, 99: 137-149.
[31] ZENG J, LIU X J, SONG L, LIN X G, ZHANG H Y, SHEN C C, CHU H Y. Nitrogen fertilization directly affects soil bacterial diversity and indirectly affects bacterial community composition., 2015, 92: 41-49.
[32] ROUSK J, BROOKES P C, BAATH E. Contrasting soil pH effects on fungal and bacterial growth suggest functional redundancy in carbon mineralization., 2009, 75(6): 1589-1596.
[33] XUN W B, HUANG T, ZHAO J, RAN W, WANG B R, SHEN Q R, ZHANG R F. Environmental conditions rather than microbial inoculum composition determine the bacterial composition, microbial biomass and enzymatic activity of reconstructed soil microbial communities., 2015, 90: 10-18.
[34] TIBBETT M, SANDERS F E. Ectomycorrhizal symbiosis can enhance plant nutrition through improved access to discrete organic nutrient patches of high resource quality., 2002, 89(6): 783.
[35] XU M G, TANG H J, YANG X Y, ZHOU SH W. Best soil managements from long-term field experiments for sustainable agriculture., 2015, 14(12): 2401-2404.
[36] HIBBETT D S. A phylogenetic overview of the Agaricomycotina., 2006, 98(6): 917.
[37] LUIS P, WALTHER G, KELLNER H, MARTIN F, BUSCOT F. Diversity of laccase genes from basidiomycetes in a forest soil., 2004, 36(7): 1025-1036.
[38] WANG J CH, SONG Y, MA T, RAZA W, LI J, HOWLAND J G. Impacts of inorganic and organic fertilization treatments on bacterial and fungal communities in a paddy soil., 2017, 112: 42-50.
[39] JANSSEN P H. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes., 2006, 72(3): 1719-1728.
[40] JONES R T, ROBESON M S, LAUBER C L, HAMADY M, KNIGHT R, FIERER N. A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses.2009, 3(4): 442.
[41] GRIFFITH R I, THOMSON B C, PHILLIP J, THOMAS B. The bacterial biogeography of British soils., 2011, 13(6): 1642-1654.
[42] FIERER N, LAUBER C L, RAMIREZ K S, ZANEVELD J, BRADFORD M A, KNIGHT R. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients., 2012, 6(5): 1007.
[43] FIERER N, BRADFORD M A, JACKSON R B. Toward an ecological classification of soil bacteria., 2007, 88(6): 1354.
[44] 曾希柏, 王亚男, 王玉忠, 林志灵, 李莲芳, 白玲玉, 苏世鸣, 沈灵凤. 不同施肥模式对设施菜地细菌群落结构及丰度的影响. 中国农业科学, 2013, 46(1): 69-79.
ZENG X B, WANG Y N, WANG Y Z, LIN Z Z, LI L F, BAI L Y, SU S M, SHEN L F. Effects of different fertilization regimes on abundance and composition of the bacterial community in greenhouse vegetable soils., 2013, 46(1): 69-79. (in Chinese)
[45] BRUUN S. Estimating turnover of soil organic carbon fractions based on radiocarbon measurements., 2005, 47(1): 99-113.
[46] FOG K. The effect of added nitrogen on the rate of decomposition of organic matter., 2010, 63(3): 433-462.
(责任编辑 李云霞)
Response and Driving Factors of Bacterial and Fungal Community to Long-Term Fertilization in Black Soil
WANG HuiYing1, XU MingGang1, ZHOU BaoKu2, MA Xiang1, DUAN YingHua1
(1Institute of Agricultural Resources and Regional Planning, Chinese Academy of Agricultural Sciences /National Engineering Laboratory for Improving Quality of Arable Land, Beijing 100081;2Soil and Fertilizer Institute, Heilongjiang Academy of Agricultural Sciences, Harbin 150086)
【Objective】This study was conducted to explore the bacterial and fungal community responses to long-term chemical fertilizer and manure application, and thus to clarify the biological mechanism of fertilization on soil fertility in black soils. It aims to provide a theoretical support for rational application of fertilizer and black soil fertility improvement. 【Method】The study was conducted on the long-term fertilization (35 years) experiment at Harbin, China. Soil samples were collected from the following four treatments: control, non-fertilization (CK), chemical nitrogen fertilizer (N), manure only (M) and M plus N (MN). Miseq pro-sequencing and qPCR technology were used to find out the difference between bacterial and fungal communities. In combination with soil properties, we analyzed the driving factors for bacterial and fungal community by multivariate statistical analysis. 【Result】Compared with CK, N treatment significantly decreased bacterial diversity and fungal diversity by 13.2%-48.5% and 4.6%-80.3%, respectively, while fungal abundance was increased by 24 times. Applied manure to N fertilization enhanced bacterial abundance and bacterial diversity by 2 times and 7.7%-46.6%, respectively. However, fungal quantity was declined by 14.2% and fungal diversity was increased by 62%-237%, comparing MN with N only fertilizer. In comparison with CK, the abundance of_,and-(bacterial classes) were significantly increased, and(fungal class) even was enhanced by 41 times with the addition of N. Compared with N treatment, the bacterial abundance kept constant for MN treatment, while the abundance of α-Proteobacteria,_andwere decreased, and_,andwere increased for M treatment. Bacterial community structure for MN and N treatments appeared similar, which were significantly different from CK and M treatments. Fungal community structure for CK, M and MN treatments were similar and significantly different from N treatment. Soil pH (6.07) and available potassium (125.5 g·kg-1) were the principal factors for the difference of bacterial community and fungal community, respectively. Soil organic matter explained both bacterial and fungal community structure alternations while the criteria was different as 28.4 g·kg-1for bacteria and 30.8 g·kg-1for fungi. 【Conclusion】Therefore, our results indicate that bacteria was sensitive to the manure, and fungi was sensitive to N fertilizer application. Long term N application stimulated the growth of acidophilic microbe, while addition of manure to N enhanced microbial diversity and promoted the growth of beneficial microorganism. Soil pH and available potassium were the principal factors driving bacterial and fungal community structure, respectively. Further detailed study is required on this aspect for the improvement of black soil quality.
bacteria; fungi; long-term fertilization; Mesiq pro-sequencing; qPCR; black soil
2017-08-07;
2017-09-25
国家重点基础研究发展计划(“973”计划)(2015CB150505)、国家重点研发计划(2016YFD0200301)、国家自然科学基金(41471247)
王慧颖,E-mail:15201530612@163.com。
段英华,E-mail:duanyinghua @caas.cn
