坏死性小肠结肠炎晚发和长病程动物模型的建立
2018-03-13王玲玲温博贤林楚琴黄堃莹江观银
王玲玲,金 芳,温博贤,冼 其,熊 慧,林楚琴,黄堃莹,江观银,黄 艳
(1.中山大学附属第六医院儿科,广东 广州 510655;2.广州市红十字会医院病理科,广东 广州 510220)
坏死性小肠结肠炎(necrotizing enterocolitis,NEC)是早产儿致死、致残的重要疾病,发病机制尚未完全阐明,临床缺乏确切有效的防治手段[1]。动物模型是研究人类疾病的基础平台,适宜的动物模型对NEC相关研究具有重要意义[2-3]。
目前公认的NEC模型是采用早产新生鼠,给予高渗配方奶人工喂养和缺氧、寒冷(或细菌内毒素LPS)刺激[Formula Feeding and Cold(or LPS)Asphyxia Stress,FFCAS(FFLAS)]进行诱导[2-4],存活时间短(平均33.6~45 h[5]),不符合人类NEC“越早产越晚发”[6]的临床特点和长时间疗效观察的实验要求。建立晚发和长病程动物模型,是NEC研究的迫切需要。
最新研究发现,NEC“越早产越晚发”与潘氏细胞损伤有关[8,20]。本研究采用双硫腙(Dithizone,Dith)选择性破坏潘氏细胞,结合FFLAS(Dith/FFLAS法),对10日龄SD大鼠进行诱导,成功建立晚发和长病程NEC模型。
1 材料与方法
1.1 实验动物与分组 10日龄SD大鼠,雌雄不拘,购自中山大学实验动物中心,随机分成4组:正常对照组(Control,n=32)采用母鼠喂养;双硫腙对照组(Dith,n=32)采用注射双硫腙+母鼠喂养;传统诱导组(FFLAS,n=32);新法建模组(Dith/FFLAS,n=47)。
1.2 晚发NEC诱导 各组乳鼠均置保育箱(T 30~31℃,H 45%~55%)。Dith组和Dith/FFLAS组腹腔注射双硫腙(Sigma-Aldrich)75 mg/kg体质量[8],Control组和FFLAS组注射等量溶媒。注射后4 h,Control组和Dith组回归母鼠喂养;FFLAS组和Dith/FFLAS组持续置保育箱,给予FFLAS刺激,具体方法参照文献[3]适当调整:插胃管注入高渗配方奶,1次/4 h,每次40 μl/g体质量;从第1次喂养开始,每12 h给予LPS(055:B5,Sigma)5 mg/(kg·次),加入配方奶中灌胃,之后置缺氧舱(容积1.4 L),予含氧5%的氮氧混合气体10 L/min通气10 min。
1.3 NEC长病程诱导 FFLAS组和Dith/FFLAS组持续给予FFLAS刺激,第7天(7 d)撤除LAS,并将FF由插胃管定量喂养改为奶嘴自主吸吮喂养,每次以半小时内拒绝3次为止。
1.4 实验终点和采样时间点 ①FFLAS组和Dith/FFLAS组于17日龄(7 d)脱离保育箱,添加面包、橙子和常规鼠饲料;所有组别均于21日龄离乳自主觅食,25日龄(15 d)结束实验。②采样时间点和样本预处理:于24 h、72 h、7 d三个时点每组随机抽取8只,以及实验终点(15 d)时所有存活鼠(Dith/FFLAS组7 d实际抽取7只,15 d为6只,其它时点及另3组各时点均为8只),麻醉处死,采集末段回肠2 cm,固定、包埋、切片。非抽样死亡(包括垂死者)亦及时解剖采样。
1.5 NEC组织学测评 肠组织切片HE染色,单人单盲光镜下观察,根据Jilling[4]标准进行评分,0:肠绒毛结构完整;1:绒毛顶端细胞脱落;2:绒毛中部结构破坏;3:绒毛完全破坏,但隐窝结构尚完整;4:隐窝破坏或透壁坏死;≥2分诊断NEC。计算各时点NEC患病率;各组损伤整体情况用(均值±标准差)表示。
1.6 潘氏细胞观察 肠组织切片AB/PAS染色标记潘氏细胞[8],光镜下观察,隐窝基部含紫红色颗粒的锥形细胞为潘氏细胞。随机观察100个结构完整的隐窝,计数潘氏细胞阳性隐窝数,以此定量表示潘氏细胞发育水平。
2 结果
2.1 临床表现 Dith/FFLAS组48~72 h普遍出现腹胀、腹泻,个别出现血便,72 h以后肉眼血便消失,但体质量仍进行性下降(见图1),撤离LAS后缓慢回升,第12天时与实验初始体质量比较差异无统计学意义(t=-1.77,P>0.05)。FFLAS组体质量增长缓慢,72 h内出现一过性大便松软,个别有轻度腹泻,但无腹胀、血便情况。Control组和Dith组无异常。

图1 时间-体质量变化百分率点线图Figure1 Plotting of weight change percentage with time-point
2.2 肠组织损伤情况和NEC患病率分析回肠典型组织学特点:Control组各时点回肠黏膜结构完整,绒毛长且排列紧密;Dith/FFLAS组24 h肠绒毛顶端上皮脱落,72 h和7 d肠黏膜充血水肿,绒毛脱落,15 d绒毛稀疏、矮短,结构紊乱。4组肠组织学评分见图2,差异均有统计学意义(F24h=3.00,F72h=17.05,F7d=14.09,F15d=4.08,P值均<0.05)。各时点均以Dith/FFLAS组评分最高,不仅与Control组差异有统计学意义(P<0.05),在72 h、7 d、15 d时点都显著高于FFLAS组和Dith组(P值均<0.05,见图2)。而且,Dith/FFLAS组不同时点病变程度差异有统计学意义(F=6.60,P<0.01);72 h和7d损伤程度显著高于24 h(q72h-24h=5.23,q7d-24h=3.66;P值均<0.05)和15 d(q72h-15d=4.84,q7d-15d=3.41;P值均<0.05)。
Control组、Dith组肠组织病理均未达到NEC诊断标准。Dith/FFLAS组24 h、72 h、7 d、15 d时点NEC患病率分别为1/8、7/8、5/7、1/6,FFLAS组则为0/8、2/8、1/8、0/8,两组患病高峰均在72 h~7 d,Dith/FFLAS组显著高于FFLAS组(P72h=0.019;P7d=0.034),Dith/FFLAS组72 h与7 d患病率差异无统计学意义(P=0.369),但显著高于24 h(P72h-24h=0.005;P7d-24h=0.034)和15 d(P72h-15d=0.016)。

图2 4组各时点回肠组织学评分Figure2 Histological scores of four groups at each time point
2.3 生存分析 截至实验终点,Control、Dith、FFLPS3组均无非抽样死亡。Dith/FFLPS组生存分析如图3,3 d存活率76.22%,7 d 58.07%,15 d(已成功离乳独立生存)38.72%。死亡高峰发生在诱导开始的第1个24 h(5只),脱离保育箱的第1个24 h死亡3只,之后未再发生死亡;24 h~7 d之间死亡的10只,有6只发生在缺氧期间。
死亡的18只乳鼠肠组织损伤情况见图4:第1个24 h NEC患病率不高(1/5),之后死亡者几乎都有不同程度的NEC样病变。
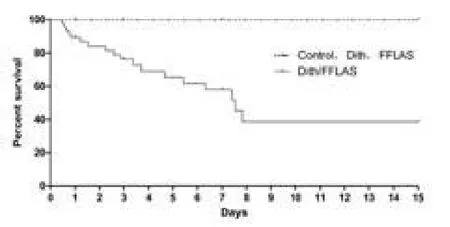
图3 生存分析Figure3 Diagram of survival analysis

图4 Dith/FFLPS组非抽样死亡乳鼠回肠组织学评分Figure4 Histological scores of pups died without sampling of group Dith/FFLPS
2.4 潘氏细胞发育情况Control组随着日龄的增长回肠潘氏细胞数量增加,AB/PAS染色增强;Dith/FFLPS组潘氏细胞发育缓慢,不仅细胞颗粒少、着色浅,而且阳性隐窝数量少。
各时点潘氏细胞发育水平见图5。4组差异均有统计学意义(F24h=7.71,F72h=6.79,F7d=13.41,F15d=16.03,P均<0.001)。Dith组和Dith/FFLPS组24 h潘氏细胞数量显著减少,Dith组72 h恢复(与Control相比,q=0.98,P>0.05)并保持正常发育;Dith/FFLPS组潘氏细胞发育缓慢,直至15 d仍显著低于Control组(P<0.05);FFLAS组发育逐渐落后,至7 d和15 d与Control组比较差异有统计学意义(q7d=4.18,q15d=4.56;P<0.05),但一直高于Dith/FFLPS组(P<0.05)。
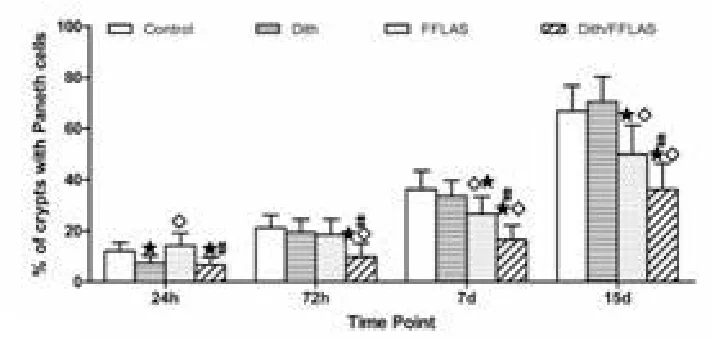
图5 4组各时点回肠组织潘氏细胞阳性隐窝数Figure5 Percentage of ileal crypts with Paneth cells at each time point
3 讨论
NEC没有自发动物模型,主要是模拟致病危险因素进行诱导[2]。目前唯一公认和成熟的NEC模型是采用早产鼠,给予高渗配方奶人工喂养,结合缺氧和寒冷刺激(FFCAS)进行诱导。该方法最初由Barlow等在1974年创立,后经多位学者改良,并将实验对象扩展到自然分娩的新生鼠(强调未进食母乳)[9],方法上省去寒冷刺激,添加细菌内毒素灌胃(FFLAS)[3]。这些对FFCAS法的合理简化,已被广泛采用[10-12]。
FFCAS(FFLAS)法建立的新生鼠NEC模型,为NEC相关研究做出了重要贡献,但存在两个问题:①与人类NEC“越早产越晚发”的特点缺乏相似性。多中心队列研究显示[6]:NEC不仅“越早产越高发”,而且“越早产越晚发”。新生鼠不仅肠道菌群尚未建立,而且消化道成熟度远低于人类[2,7],用于发病机制研究时,在某些方面可能会“失真”。②不能进行长时间观察,在治疗研究方面缺乏适用性。新生鼠FFCAS法诱导24 h后开始陆续发病[4],72~96 h成模率52.7%~85%[13-15],平均存活时间33.6~45 h[5,16]。为了有足够的观察期,目前的治疗学研究都是给药与造模同时进行,本质上属于预防性干预。成模后干预的报道只有一篇,仅观察了4 d[17]。因此,建立晚发和长病程动物模型,是NEC研究的迫切需要。
如果新生鼠出生一段时间后再诱导建模,不仅能满足“晚发”的要求,而且对疾病的耐受能力增强,或可带病存活较长时间从而建立长病程模型。但是,由于母乳的保护作用,以及随着日龄的增长肠黏膜自稳能力日趋成熟,FFCAS(FFLAS)法能否诱导成功是个问题。研究发现,母鼠喂养12~24 h的新生小鼠,NEC发病率明显低于母鼠喂养不足12 h者(44%vs 70%,P<0.05)[5];小鼠出生14 d后FFCAS法就不能诱导出NEC[2]。
FFLAS法在新生大鼠的72~96 h成模率为50%~75%[3],尚未见用于较大日龄大鼠。本项目结果显示,10日龄SD大鼠在FFLAS诱导下体质量增长缓慢,临床无典型NEC症状;72 h成模率(2/8)。结合双硫腙进行诱导(Dith/FFLAS法),则普遍出现NEC临床症状,72 h成模率87.5%(7/8),两者的成模率和损伤程度差异有统计学意义(P<0.05)。由此可见,随着乳鼠日龄的增加,FFLAS不再适宜的建模方法,Dith/FFLAS能有效建立晚发NEC模型。
Dith是重金属螯合剂,能选择性破坏潘氏细胞(富含锌)。新生鼠潘氏细胞尚未发育,随着日龄的增加逐渐成熟。Zhang等[8]采用14~16日龄小鼠,腹腔注射Dith,继之克雷伯杆菌灌胃(Dith/Kleb),16 h NEC成模率76.67%;单纯Dith注射或细菌灌胃都不能诱导NEC样病变。类似的方法在5日龄小鼠[8]和4日龄大鼠[19]都不能诱导NEC。这些研究提示:NEC发生在潘氏细胞相对成熟之后[8,20]。
潘氏细胞是肠黏膜最重要的天然免疫细胞,它不仅产生丰富的抗菌肽(AMP),而且产生多种促炎因子(TNF-a、IL-1β、IL-17等)[7,20]。
人类早产儿潘氏细胞的数量和功能远低于足月儿。NEC“越早产越高发”和“越早产越晚发”,这一临床现象提示:NEC患儿肠黏膜免疫功能不成熟,但又要有一定的成熟度,“过度炎症反应”才可能发生[7]。结合上述动物实验研究,有学者推测:早产儿生后需要一段时间,待潘氏细胞发育至一定的成熟度,有足够的促炎因子和反应能力的情况下,才能启动NEC[7,20]。
基于上述认识,2013年McElroy等[20]提出了NEC源自潘氏细胞损伤的“自底而上”假说(‘bottom up’hypothesis):微生物或其毒素入侵肠隐窝,破坏潘氏细胞;受损的潘氏细胞从基底侧向黏膜固有层及微血管释放TNF-a、IL-1β、IL-17等促炎因引发炎症反应和血管内皮损伤、血栓形成,导致肠黏膜凝固性坏死。
本文注射Dith的两组乳鼠24 h潘氏细胞数量均显著减少,但Dith/FFLAS组24 h成模率很低,72 h和7 d成模率和病损严重程度最高;对死亡乳鼠的病理分析与这一趋势吻合。Dith组和FFLAS组均无死亡,Dith组无NEC样病变,FFLAS组各时点患病率和病损程度均显著低于Dith/FFLAS组,说明Dith/FFLAS组死亡和成模都不是Dith或FFLAS单一作用的结果,而是两者的协同效应。
但死亡峰先于成模峰,死亡峰在24 h内,这一时间段不论死亡还是抽样的乳鼠NEC患病率都非常低(图2、图3)。这一现象与上述Dith/Kleb法16 h成模的实验结果不同,不支持“自底而上”假说,但也不能否定这一假说,因为LPS不能模拟活菌的生物学行为,而且受试对象(大鼠vs小鼠)和发育水平(日龄)也不同。
对于Dith/FFLAS法能在10日龄SD大鼠成功诱导NEC,以及死亡峰先于成模峰的原因,我们推测:①Dith对潘氏细胞的破坏削弱了肠屏障功能,使得更多的LPS进入血循环,加重毒血症程度;②Dith使金属酶功能障碍,削弱了机体对毒血症和缺氧的耐受能力;③金属酶功能和肠屏障功能障碍增加了机体的NEC易感性。
目前尚未见对NEC“长病程”模型进行系统研究的报道。Zani等[11]在进行干细胞治疗NEC的实验研究时,为了有足够的疗效观察期,对FFLAS法作了调整:给予新生大鼠FFLAS刺激,48 h撤除LPS,96 h停止缺氧刺激,单纯给予高渗配方奶人工喂养(FF),第7天存活率不足20%,没有NEC患病率的描述。提前撤除诱导因素可能降低患病率,以此为代价谋求延长疗效观察时间,反映了传统NEC模型的局限性。
本项目Dith/FFLAS法诱导第7天NEC患病率71.43%(5/7),第15天病变仍未完全修复(图2),平均存活时间6.25天,说明本项目成功建立了长病程NEC模型。
本文Dith组体质量正常增长,72 h潘氏细胞数量恢复正常,提示此时Dith对机体的直接影响已经消失。而且,FFLAS组7 d组织学评分与Control组比较差异无统计学意义(见图2)。说明长病程NEC不是Dith或FFLAS直接所致,而是在Dith/FFLAS诱导成模后的病变迁延,是FFLAS阻碍肠黏膜修复的结果。
肠黏膜修复的主体是肠上皮干细胞(Intestinal Sterm Cell,ISC),与潘氏细胞紧密相邻;潘氏细胞是其“卫士”和“龛细胞”,对维持肠上皮稳态有重要意义[21]。本文不仅Dith/FFLAS组潘氏细胞恢复缓慢,而且FFLAS组潘氏细胞发育也明显落后,这与Estienne等[22]的研究结果一致,他们发现早期母爱剥夺使乳鼠潘氏细胞数量减少,功能发育迟滞。而且,细菌内毒素LPS抑制肠上皮修复[18]。LPS是TLR4的配体。最新研究发现:ISC表达TLR4,激活TLR4诱导ISC凋亡,导致未成熟小鼠NEC易感性增加和肠黏膜修复能力下降[23]。
因此,Dith/FFLAS法建立NEC长病程模型的机制可能是:Dith破坏潘氏细胞,人工喂养造成的母爱剥夺使之发育延迟,直接影响ISC生存的微环境,而且削弱了肠黏膜免疫屏障,使得肠腔微生物成分(如LPS)更易接触到ISC,与其表面的TLR4结合,激活ISC凋亡程序,从而削弱肠黏膜修复能力,使NEC病变迁延。撤除LPS后体质量增长也支持这一推断。
综上所述,本文结果说明:①传统的FFLAS法在10日龄大鼠的成模率低,不适合作为晚发NEC的建模方法;②Dith/FFLAS能成功建立晚发NEC模型,成模高峰为72 h,成模率87.50%;③在晚发NEC的基础上继续给予FFLAS刺激,能成功诱导NEC长病程模型,病变至少迁延至第7天,第15天仍未完全修复;④38.72%的模型鼠能成功离乳独立生存,观察期可任意延长。
本文首次对NEC长病程模型进行了初步系统的探索,率先用Dith/FFLAS法建立晚发和长病程模型,为NEC相关研究提供新平台。本模型尤其适合治疗学研究。
[1] Schnabl KL,VanAerde JE,Thomson AB,et al.Necrotizing enterocolitis:A multifactorial disease with no cure[J].World J Gastroenterol,2008,14(14):2142-2161.
[2] Sodhi C,Richardson W,Gribar S,et al.The development of animal models for the study of necrotizing enterocolitis[J].Disease Models&Mechanisms,2008,1(2-3):94-98.
[3] Zani A,Cordischi L,Cananzi M,et al.Assessment of a Neonatal Rat Model of Necrotizing Enterocolitis[J].Eur J Pediatr Surg,2008,18(6):423-426.
[4] Jilling T, Lu J, Jackson M, et al. Intestinal EpithelialApoptosis Initiates Gross Bowel Necrosis in an Experimental Rat Model of Neonatal Necrotizing Enterocolitis[J]. Pediatr Res, 2004,55(4):622-629.
[5] Tian R,Liu SXL,Williams C,et al.Characterization of a necrotizing enterocolitis model in newborn mice[J].Int J Clin Exp Med,2010,3(4):293-302.
[6] Yee WH,Soraisham AS,Shah VS,et al.Canadian Neonatal Network:Incidenceandtiming of presentation of necrotizing enterocolitis in preterm infants[J].Pediatrics,2012,129:e298-e304.
[7] Underwood MA.Paneth cells and necrotizing enterocolitis[J].Gut Microbes,2012,3(6):562-565.
[8] Zhang C,Sherman MP,Prince LS,et al.Paneth cell ablationin the presence of Klebsiellapneumoniae induces necrotizing enterocolitis(NEC)-like injury in the small intestine of immature mice[J].Dis Mod-el Mech,2012,5:522-532.
[9] Nadler EP,Dickinson E,Knisely A,et al.Expression of inducible nitricoxide synthase and interleukin-12 in experimental necrotizing enterocolitis[J].J Surg Res,2000,92(1):71-77.
[10]Halpern MD,Khailova L,Dania MH,et al.Decreased development of necrotizing enterocolitis in IL-18-deficient mice[J].Am J Physiol Gastrointest Liver Physiol,2008,294:G20-G26.
[11]Zani A,Cananzi M,Fascetti-Leon F,et al.Amniotic fiuid stem cells improve survival and enhance repair of damaged intestine in necrotizing enterocolitis via a COX-2 dependent mechanism[J].Gut,2014,63(2):300-309.
[12]Underwood MA,Kananurak A,Coursodon CF,et al.Bifidobacterium bifidum in a rat model of necrotizingenterocolitis:antimicrobial peptideand protein responses[J].Pediatr Res,2012,71(5):546-551.
[13]Deplaen IG,Liu SXL,Tian R,et al.Inhibition of Nuclear Factor-kB Ameliorates Bowel Injury and Prolongs Survival in a Neonatal Rat Model of Necrotizing Enterocolitis[J].PediatrRes,2007,61(6):716-721.
[14]Caplan MS,Hedlund E,Adler L,et al.Role of asphyxia and feeding in a neonatal rat model of necrotizing enterocolitis[J]. Pediatric Pathology,1994,14(6):1017-1028.
[15]Jilling T,Simon D,Lu J,et al.The roles of bacteria and TLR4 in rat and murine models of necrotizingenterocolitis[J].JImmunol,2006,177(5):3273-3282.
[16]Yang JX,Watkins D,Chen CL,et al.Heparin-binding epidermal growth factor-Like growth factor and Mesenchymal stem cells act synergistically to prevent experimental necrotizing enterocolitis[J]. J Am Coll Surg,2012,215(4):534-545.
[17]Tayman C,Uckan D,Kilic E,et al.Mesenchymal stem cell therapy in necrotizing enterocolitis: A rat study[J].Pediatr Res,2011,70(5):489-494.
[18]Leaphart CL,Cavallo J,Gribar SC,et al.A Critical Role for TLR4 in the Pathogenesis of Necrotizing Enterocolitis by Modulating Intestinal Injury and Repair[J].J Immunol,2007,179(7):4808-4820.
[19]ShermanMP,BennettSH,HwangFF,etal.Paneth cells and antibacterial host defense in neonatal smallintestine[J].InfectImmun,2005,73(9):6143-6146.
[20]McElroy SJ,Underwood MA,Sherman MP.Paneth Cells and Necrotizing Enterocolitis:A Novel Hypothesis forDisease Pathogenesis[J].Neonatology,2013,103(1):10-20.
[21]Sato T,van Es JH,Snippert HJ,et al.Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts[J].Nature,2011,469(7330):415-418.
[22]Estienne M,Claustre J,Clain-Gardechaux G,et al.Maternal deprivation alters epithelial secretory cell lineages in rat duodenum:role of CRF-related peptides[J].Gut,2010,59(6):744-751.
[23]Afrazi A,Branca MF,Sodhi CP,et al.Toll Like Receptor 4-mediated Endoplasmic Reticulum Stress in Intestinal Crypts Induces Necrotizing Enterocolitis[J].J Biol Chem,2014,289(14):9584-9599.
