Deoxygedunin对D-半乳糖联合AlCl3诱导大鼠阿尔茨海默病病理性改变的影响*
2018-03-13陈建国江祺川王若雅吴亚更
陈建国, 江祺川, 温 博, 王若雅, 吴亚更, 李 响
(锦州医科大学附属第一医院头颈外科, 辽宁 锦州 121001)
阿尔茨海默病(Alzheimer's disease,AD)是最常见的神经退行性疾病之一,以渐进性认知功能障碍和行为损害为特征[1]。虽然老年斑和神经原纤维缠结(neurofibrillary tangles,NFT)通常被认为是AD的标志,氧化应激和突触功能障碍也与认知功能下降相关[2, 3]。β淀粉样蛋白(amyloid protein,Aβ)来源于淀粉样前体蛋白(amyloid precursor protein,APP)淀粉样变性过程,是AD病理生理学中老年斑的主要成分。Aβ在AD的发病机制中起着重要作用,并且在通过直接和间接的方式产生自由基的可溶性Aβ低聚物中发挥重要作用[4]。NFT由过度磷酸化的tau蛋白聚集成对螺旋状细丝而形成。因此,抑制Aβ聚积和tau蛋白过度磷酸化已被认为是AD的治疗策略。
因AD的病因及发病机制较为复杂,因此针对该疾病的动物模型的制备也多种多样,主要包括自然衰老模型、Aβ 注射造模、D-半乳糖造模、转基因模型、胆碱能系统损伤模型以及中毒损伤模型等,本研究所采用模型属于多重复制模型,接近于自然衰老模型,且重现Aβ沉积的老年斑、Tau蛋白过度磷酸化及神经原纤维缠结等AD经典病理变化,能较好地模拟 AD 的相关衰老的表型,具有操作简单、价格低、重复性高等特点。
脑源性神经生长因子(brain derived neurotrophic factor,BDNF)对神经元的存活和神经突的生长具有重要作用,BDNF参与学习和记忆过程,AD患者的血清BDNF水平较正常受试者降低,同样AD转基因小鼠BDNF表达下调,证明BDNF在AD的发生和发展中发挥重要作用[5, 6]。BDNF通过活化其细胞膜上的TrkB受体发挥其生物学作用。
Deoxygedunin是一种新型的,高亲和力的TrkB受体激动剂,模拟BDNF与TrkB结合,使TrkB受体发生自体磷酸化,进一步激活神经保护作用的分子通路而发挥生物学作用。Deoxygedunin具有抗氧化、抑制神经元死亡、改善学习记忆、改善抑郁和促进神经再生等作用。有研究显示TrkB受体激动剂Deoxygedunin对1-甲基-4-苯基-1, 2, 3, 6-四羟吡啶诱导的帕金森疾病模型的多巴胺能神经元具有保护作用[7]。但目前关于Deoxygedunin是否对神经退行性疾病AD具有神经保护作用,以及该神经保护作用是通过何种机制来发挥尚未有报道,因此本实验通过评估Deoxygedunin对D-半乳糖联合AlCl3诱导的阿尔茨海默病大鼠是否具有神经保护作用以及探讨其机制,以期为AD的治疗提供新的神经保护策略。
1 材料与方法
1.1 实验动物与分组
雄性SD大鼠购于北京军事医学科学院实验动物中心,体重(250±20) g,SD大鼠许可证号:SCXK-(军)2012-0004。大鼠饲养环境为SPF级别,温度为25℃昼夜循环交替。将大鼠分为3组(n=12):对照组(Control)、模型组(AD)和干预组(AD+Deo)。模型组采用D-半乳糖180 mg/(kg·d)腹腔注射联合AlCl315 mg/(kg·d)灌胃,连续给药12周,构建阿尔茨海默病大鼠模型;对照组腹腔注射和灌胃等体积生理盐水;Deo组是在造模12周后,给予Deoxygedunin 5 mg/(kg·d)腹腔注射,连续2周。
1.2 主要抗体、试剂和仪器
用于蛋白免疫印迹实验的兔单克隆抗体Anti-Tau(磷酸化T231位点)、 TrkB/p-TrkB、AKT/p-AKT、ERK1/p-ERK1抗体均购自英国Abcam公司;辣根过氧化物酶标记的羊抗兔二抗购自CST公司;Deoxygedunin购自Gaia公司;超氧化物歧化酶(superoxide dismutase,SOD),谷胱甘肽过氧化物酶(glutathione peroxidase,GSH-Px),谷胱甘肽(glutathione,GSH)和丙二醛(malondialdehyde,MDA)来自碧云天生物技术公司;AlCl3购自国药集团化学试剂有限公司;Biotin-WO2来自德国Abeta有限公司;辣根过氧化物酶-亲和素购自美国Pierce公司;TMP 微孔过氧化物酶系统购自美国Kirkegaard和Perry实验室。
1.3 水迷宫实验检测大鼠的空间学习记忆能力
Deo给药结束前5 d行Morris水迷宫实验。通过挂在泳池周围的图案训练大鼠寻找隐匿平台。将大鼠从不同象限放入泳池,记录大鼠到达平台的时间,即逃避潜伏期。若60 s内大鼠仍未到达平台,将其引导到平台上停留5 s,并将潜伏期记录为60 s,每只大鼠接受连续6 d的训练,4次/天(n=12)。第7日撤去水上平台,固定入水点,记录大鼠60 s内在目的象限内停留的时间、穿越平台次数和游泳速度,用于评估大鼠学习和记忆能力。
1.4 ELISA实验测量Aβ40和Aβ42蛋白的浓度
水迷宫实验结束2 h后,取大鼠双侧新鲜海马组织,采用双抗体夹心法酶联免疫吸附试验(ELISA)实验定量分析各组大鼠海马Aβ水平(n=4)。用特异性单克隆抗体结合样品中的Aβ40和Aβ42蛋白,然后用Biotin-WO2抗体探测,然后用辣根过氧化物酶-亲和素进行结合。使用TMP微孔过氧化物酶系统检测辣根过氧化物酶活性。严格按 ELISA 试剂盒的操作规范进行试验,所有标本和试剂盒使用前均提前0.5 h放在室温平衡。
1.5 Western blot法检测TrkB信号通路上ERK1、AKT和TrkB的蛋白表达
水迷宫结束后2 h处死大鼠取材,取双侧新鲜海马置于匀浆液中进行组织匀浆(n=4)。使用BCA方法测量蛋白质浓度。将处理后的蛋白样本通过10%SDS-聚丙烯酰胺凝胶电泳分离等量的蛋白质并转移至PVDF膜。5% 脱脂奶粉封闭 2 h,一抗孵育 4℃ 过夜(TrkB/pTyr516-TrkB、Akt/pSer473-Akt、ERK1/p-ERK1稀释比例为1∶10 000)。然后与辣根过氧化物酶标记的二抗室温避光温育2 h,并用化学发光底物试剂盒显色。使用ImageJ软件对蛋白免疫印迹条带进行扫描和分析。
1.6 免疫组织化学染色法检测大鼠脑皮质Aβ1-40和Aβ1-42 蛋白表达情况
Morris水迷宫认知能力测试结束2 h后,大鼠行4%多聚甲醛心脏灌流取材,用4%多聚甲醛固定48 h,进行切片染色(n=4)。具体步骤如下:室温脱蜡、水化;采用微波修复抗原;免疫组化笔画圈;3%过氧化氢灭活内源酶活性;封闭非特异性位点;甩掉封闭液,滴加一抗Anti-Tau (磷酸化T231位点)覆盖组织,然后放入湿盒内4℃过夜。(稀释比例1∶200);次日4℃冰箱取出湿盒,室温复温30 min,滴加二抗;滴加链霉菌抗生物素-过氧化物酶溶液;滴加DAB溶液;苏木素复染。显微镜下观察大脑皮质Aβ1-40和Aβ1-42 表达情况。
1.7 统计学处理
2 结果
2.1 Deoxygedunin对D-半乳糖联合AlCl3诱导的大鼠学习和记忆缺陷的影响
Morris水迷宫实验用来测试大鼠的学习和记忆能力。与对照组相比,模型组大鼠逃避潜伏期显著增加;与模型组相比,Deo干预组逃避潜伏期显著降低,尤其是第7日降低最为明显(表1)。与对照组相比,模型组大鼠穿越平台次数较少;与模型组相比,Deo干预组大鼠穿越平台次数显著增多(表2)。所有各组大鼠的游泳速度没有显著差异(表2)。这些结果表明Deoxygedunin干预有效地恢复了D-半乳糖联合AlCl3诱导的学习和记忆功能缺陷。

Tab.1 The latency of time to find platform of the three groups(s, n=12)
AD: Alzheimer's disease model group; AD+Deo: Alzheimer's disease model+deoxygedunin intervention group
*P<0.05,**P<0.01vscontrol group;#P<0.05,##P<0.01vsmodel group


GroupNumber of crossings(n/s)Swimming speed(cm/s)Control4.40±0.9417.53±2.23AD2.17±0.92∗∗16.03±1.83AD+Deo3.87±0.60##19.00±2.94
**P<0.01vscontrol group;##P<0.01vsmodel group
2.2 Deoxygedunin对D-半乳糖联合AlCl3诱导的Aβ 积聚的影响
与对照组相比,发现D-半乳糖联合AlCl3的应用显著增加海马Aβ集聚;与模型组相比,Deoxygedunin应用会减少Aβ40和Aβ42的表达水平(表3)。
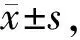

GroupAβ40Aβ42Control25.52±1.7110.50±1.29AD70.00±5.71∗∗47.00±3.27∗∗AD+Deo30.03±2.87##18.03±2.19##
**P<0.01vscontrol group;##P<0.01vsmodel group
2.3 Deoxygedunin对D-半乳糖联合AlCl3诱导的氧化应激反应的影响
通过测定海马中MDA和GSH水平以及SOD活性来测定氧化应激。D-半乳糖联合AlCl3处理可增加MDA水平,同时抑制GSH水平和SOD活性(表4)。Deoxygedunin可以恢复D-半乳糖联合AlCl3诱导的这些效应。MDA代表脂质氧化,GSH-Px活性表明消除自由基的能力,Deoxygedunin可改善脂质氧化和清除自由基的能力。


GroupMDA(nmol/mg)GSH-Px(pg/mg)SOD(U/mg)Control0.91±0.129.98±1.393.90±0.75AD1.48±0.16∗∗6.03±0.69∗∗1.40±0.43∗∗AD+Deo1.08±0.17#10.03±0.82 ##3.30±0.42##
AD: Alzheimer's disease model group; AD+Deo: Alzhei mer's disease model+deoxygedunin intervention group
**P<0.01vscontrol group;#P<0.05,##P<0.01vsmodel group
2.4 Deoxygedunin对Tau蛋白过度磷酸化的影响
Tau蛋白在大脑皮质神经元内呈棕黄色阳性着色。对照组大鼠大脑皮质神经元胞膜上有少量淡黄色阳性颗粒。模型组大鼠脑皮质神经元胞膜染色呈棕黄色强阳性。Deo组大鼠脑皮质神经元胞膜含有弱阳性浅棕黄色颗粒(图1,见彩图页Ⅰ)。
2.5 Deoxygedunin对TrkB信号通路上ERK1、AKT和TrkB的蛋白表达的影响
BDNF/TrkB通路对突触和神经元的可塑性均具有利作用。Deoxygedunin是一种新型TrkB激动剂,能起到神经保护作用。用Deoxygedunin干预的大鼠海马中TrkB的磷酸化水平降低。BDNF/TrkB通路的重要下游分子TrkB,AKT和ERK1活性检测结果显示Deoxygedunin可逆转被D-半乳糖联合AlCl3抑制的TrkB信号转导通路(图2,表5)。

Fig.2The expressions of pTyr516-TrkB, pSer473-Akt and pERK1 protein detected with Western blot


GrouppTyr516-TrkB/TrkBpSer473-Akt/AktpERK1/ERK1Control1.00±0.111.00±0.231.00±0.19AD0.40±0.03∗0.49±0.05∗0.23±0.11∗AD+Deo1.13±0.26#0.96±0.20#1.03±0.27 #
*P<0.05vscontrol group;#P<0.05vsmodel group
3 讨论
AD发病机制极其复杂,其典型的病理特征是老年斑形成和神经纤维缠结。因此,改善Aβ毒性、抑制Aβ表达或抑制Tau蛋白过度磷酸化是防治AD的一个重要切入点[8, 9]。本研究的目的是探讨黄酮类化合物Deoxygedunin在D-半乳糖联合AlCl3诱导AD样病理改变所致功能障碍的保护作用。本研究通过D-半乳糖联合AlCl3制备AD动物模型,其中D-半乳糖的代谢物因不能被机体代谢而堆积在细胞内,导致细胞代谢功能紊乱,产生氧化损伤,造成神经元结构和功能的变化而出现类似 AD 疾病的表型,AlCl3可刺激氧自由基产生,增强脂质过氧化,促使Aβ沉积、寡聚[10, 11]。因此,该模型可以作为筛选AD潜在的治疗药物的动物模型。本研究采用D-半乳糖180 mg/(kg·d)腹腔注射联合AlCl315 mg/(kg·d)灌胃,连续给药12周,构建阿尔茨海默病大鼠模型。给药12周后大鼠海马组织内出现Aβ沉积和Tau蛋白表达过度增加,证实D-半乳糖联合AlCl3可成功复制阿尔茨海默病大鼠模型。
有证据表明AD患者的BDNF-TrkB系统紊乱影响海马依赖性记忆和认知功能障碍[12, 13]。BDNF通过活化其细胞膜上的TrkB受体发挥其生物学作用,但作为大分子的BDNF并不能穿过血脑屏障。因此,寻找小分子BDNF类似物是激活BDNF-TrkB通路发挥神经保护作用的关键所在Deoxygedunin是从印度苦楝树中提取而来的一种黄酮类物质,具有抗氧化、抗肿瘤和改善学习记忆的作用[14]。Deoxygedunin是一种小分子BDNF的类似物,可通过血脑屏障,刺激BDNF表达并激活其特异性受体TrkB,被期望能发挥神经保护作用。
Aβ在AD中的神经毒性作用已被广泛研究,包括损害突触可塑性,诱导凋亡,促进Tau磷酸化和氧化应激。有研究表明,D-半乳糖联合三氯化铝可诱导小鼠脑组织内出现Aβ沉积和氧化应激等病理特征[15]。因此,本研究还检测了Deoxygedunin干预后大鼠脑组织中Aβ沉积、Tau蛋白磷酸化和氧化应激的变化,结果发现Deoxygedunin可逆转D-半乳糖联合AlCl3诱导的Aβ积聚、Tau蛋白的过度磷酸化和氧化应激反应。本研究结果表明,Deoxygedunin可抵抗D-半乳糖和三氯化铝神经毒性而发挥保护作用。
体内外研究还发现Deoxygedunin是一种较强的TrkB受体激动剂[16],它可直接激活大脑TrkB受体,TrkB被激活后刺激信号转导通路如PI3K/Akt,Akt是调节CREB活化的重要激酶,并且反过来又修饰学习和记忆所必需的分子表达。有研究显示,AD转基因小鼠大脑中Akt活性降低,表现为磷酸化水平下降[17, 18]。ERK是Tau蛋白磷酸化和细胞存活的重要激酶,它可因MEK/ERK信号转导通路中TrkB的活化而被激活[19]。在先前的研究中已报道过Deoxygedunin的神经保护作用[7],但大多数研究都集中在抑郁样行为或改善学习记忆和认知功能[6]。本研究旨在探讨Deoxygedunin对AD的神经保护作用。Western blot实验显示用Deoxygedunin干预的大鼠的海马中,TrkB的磷酸化水平降低。我们进一步检测了BDNF/TrkB通路的重要下游分子TrkB,AKT和ERK1的活性,结果发现Deoxygedunin可逆转被D-半乳糖联合AlCl3抑制的TrkB信号转导通路。水迷宫结果也显示,与模型组相比,Deo组大鼠寻找隐匿平台的潜伏期显著降低,穿越平台的次数和目标象限停留的时间均显著增多。本研究结果表明,补充模拟BDNF特异性作用的TrkB激动剂Deoxygedunin可体内激活TrkB,从而改善D-半乳糖联合AlCl3诱导的认知功能障碍包括学习记忆障碍。
综上所述,Deoxygedunin可改善D-半乳糖联合AlCl3诱导的大鼠AD样学习记忆障碍,增加脑组织SOD和GSH-Px的活性、降低脑组织MDA水平以及改善脑组织Aβ积聚和tau蛋白异常磷酸化。其具体作用机制可能是通过激活TrkB受体,提高下游信号通路的pTyr516-TrkB /TrkB、pSer473-Akt/Akt和pERK/ERK的磷酸化水平,激活TrkB信号通路。本研究提示Deoxygedunin可通过激活TrkB信号通路来发挥其神经保护作用,Deoxygedunin可作为潜在的AD靶向药物,为AD防治提供了新策略,但向临床转化及应用仍需要大量的关于药代动力学和生物利用度相关研究。
