吉祥草配伍余甘子的提取工艺研究
2018-03-06黄兴文黄诗娅
王 鹤 李 江 黄兴文 黄诗娅
贵阳中医学院,贵州 贵阳 550002
吉祥草与余甘子均为贵州十大苗药,药用资源广泛。吉祥草为百合科植物Reineckeacarnea(Aner.) Kunth的干燥全草[1-3],在贵州、湖南、广西等苗族人口主要聚集区使用较为广泛[4],具有滋阴润肺、凉血止血、解毒利咽之功效,用于肺燥咳喘,阴虚咳嗽,咳血,治疗肺热咳嗽,跌打损伤,疮毒等[5]。余甘子为大戟科叶下珠属(Phyllanthus)植物余甘子PhyllanthusemblicaL.的干燥成熟果实[6],系藏、苗族民间常用药[7],具有清热凉血,消食健胃,生津止渴之功效。用于血热血瘀,消化不良,腹胀,咳嗽,喉痛,口干。吉祥草长于解毒清肺止咳,余甘子长于清热润肺化痰,两药相需配伍共奏清热解毒、润肺止咳化痰之功。课题组调查发现民间将两药配伍应用,用于治疗肺癌具有较好的疗效。为了进一步提高两药配伍疗效,实验建立总皂苷、总黄酮、没食子酸的含量测定方法,采用正交设计实验优化吉祥草配伍余甘子提取工艺。
1 仪器与材料
1.1 仪器 日立Primaide高效液相色谱仪(天美科学仪器有限公司); JA2003N电子分析天平;721型可见分光光度计。
1.2 材料 吉祥草采于贵州六枝县,经贵阳中医学院生药教研室孙庆文教授鉴定为百合科吉祥草属植物吉祥草Reineckiacarnea(Andr.)Kunth 的带根全草;余甘子产地为贵州晴隆县,药材经贵阳中医学院生药教研室孙庆文教授鉴定为大戟科叶下珠属(Phyllanthus)植物余甘子PhyllanthusemblicaL.的成熟果实。吉祥草、余甘子对照药材购自中国药品生物制品鉴定所;薯蓣皂苷购自成都曼斯特生物科技有限公司;没食子酸购自中国药品生物制品鉴定所;芦丁对照品购自成都瑞芬思生物科技有限公司。
2 方法及结果
2.1 总皂苷含量测定[8]
2.1.1 供试品溶液的制备 精密称定配伍药材(吉祥草∶余甘子=2∶1)粗粉5.0 g,甲醇回流提取4 h,回收甲醇,加入蒸馏水使之溶解,滤过,滤液转移至100 mL的容量瓶中加蒸馏水稀释至刻度,摇匀。准确吸取10 mL于分液漏斗中,用石油醚分6次萃取,每次用量分别为15、10、10、10、10、10 mL,取水层用水饱合正丁醇溶液萃取4次,每次用量分别为15、10、10、10 mL,合并正丁醇萃取液并用正丁醇饱和水溶液20 mL洗涤,回收正丁醇液至干,残渣用甲醇溶解并定容于25 mL容量瓶中,作为供试品溶液。
2.1.2 对照品溶液的制备 精密称取薯蓣皂苷对照品(干燥至恒重)1.21 mg,置于10 mL容量瓶中,加甲醇溶解并定容至刻度,摇匀,作为对照品溶液(121 μg/mL)。
2.1.3 测定波长的选择 分别精密吸取对照品溶液1.5 mL、供试品溶液0.6 mL置于具塞试管中,挥干甲醇,再分别加入5%的香草醛冰醋酸溶液0.2 mL和高氯酸0.8 mL,摇匀,密塞,于60℃水浴显色15 min,取出后立即用冰水冷却5 min,再加入5 mL冰醋酸稀释,摇匀,静置10 min,在200~900nm进行扫描,结果如图1、图2所示。两者均在460 nm处,有最大吸收峰。
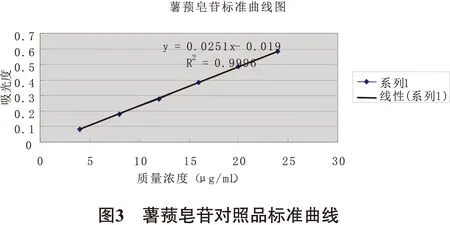
2.1.4 线性关系考察 分别精密移取薯蓣皂苷对照品溶液0.2、0.4、0.6、0.8、1.0、1.2 mL置于具塞试管中,挥干甲醇,再分别加入5%的香草醛冰醋酸溶液0.2 mL和高氯酸0.8 mL,摇匀,密塞,60℃水浴显色15 min,取出后立即用冰水冷却5 min,再加入5 mL冰醋酸稀释,摇匀,静置10 min,于460 nm处测定其吸光度。以吸光度(A)为纵坐标,质量浓度(C)为横坐标进行线性回归,回归方程为:y=0.0251x-0.0190(R=0.9996,n=6),结果表明:薯蓣皂苷在4~24 μg/mL浓度范围与吸光度成良好的线性关系,如图3所示。
2.1.5 精密度试验 吸取对照液0.8 mL,共6份,按2.1.3项下的操作方法显色,于460 nm处测定吸光度,计算RSD=1.71%,结果表明方法的精密度良好,见表1。
2.1.6 稳定性试验 精密吸取样品溶液0.4 mL,按2.1.3项下的操作方法显色,于460 nm处分别在0、15、30、45、60、90、120 min测定吸光度,结果表明样品液显色后在120 min内稳定,RSD=0.86%,结果见表1。2.1.7 重复性试验 精密称取同一批配伍药材6份,每份5g,按2.1项下方法制备供试品溶液,另取供试品溶液0.4 mL按2.1.3项下操作方法显色,于460 nm处测定吸光度,并计算含量,结果平均含量为1.40%,RSD=1.50%。结果表明方法的重复性良好。
2.1.8 加样回收试验 精密称取2.1.7项下已知含量的同一批次配伍药材2.5g,共6份,分别加入等量薯蓣皂苷对照品适量,按2.1.3项下的操作方法显色,于460 nm处测定吸光度,结果见表2。
2.2 没食子酸含量测定[9-10]
2.2.1 色谱条件 色谱柱:Venusil XBP C18(150 mm×4.6 mm,5 μm);流动相:甲醇:0.3%磷酸水(4∶96);柱温:30℃;流速:1 mL/min;检测波长:273 nm。理论塔板数按没食子酸峰计算,应不低于2000。

表1 稳定性试验考察结果
表2 加样回收率试验结果
2.2.2 供试品溶液的制备 精密称定配伍药材(吉祥草:余甘子=2∶1)粗粉0.2 g,置具塞锥形瓶中,精密加入50%甲醇50 mL,称定重量,回流1 h,放冷,再称定重量,用50%甲醇补足减失的重量,摇匀,滤过,过0.45 μm微孔滤膜,作为供试品溶液。2.2.3 对照品溶液的制备 精密称取没食子酸对照品0.026 20 g,置于25 mL容量瓶中,用50%甲醇溶解并稀释至刻度,摇匀。精密吸取0.7 mL上述溶液于10 mL容量瓶中,用50%甲醇稀释至刻度,摇匀,即得每1 mL含73.36 μg的对照品溶液。
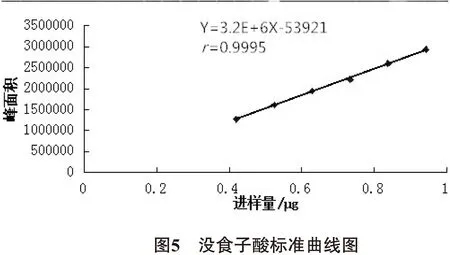
2.2.4 线性关系考察 精密吸取上述溶液0.2、0.4、0.5、0.6、0.7、0.8、0.9 mL分别置于10 mL容量瓶中,用50%甲醇稀释至刻度,摇匀。按上述色谱条件,进样体积10μL,测定没食子酸峰面积。其HPLC出峰情况见图4。以没食子酸峰面积值Y为纵坐标,进样量X(μg)为横坐标,求得没食子酸标准曲线回归方程和线性相关系数分别为Y=3 159.3X-53 921(r=0.999 5),结果表明没食子酸进样量在0.419 2~0.943 2 μg内线性关系良好,结果如图5所示。

2.2.5 精密度试验 精密吸取对照品溶液,连续重复进样6次,每次10 μL,记录峰面积,结果表明,RSD为1.39,小于3%,表明仪器的精密度良好。
2.2.6 稳定性试验 取供试品溶液,在上述色谱条件下,分别于0、1、3、6、9、12 h时进样,记录峰面积,结果表明,峰面积的RSD值为0.98%,表明样品溶液在室温条件下12 h内基本稳定。结果见表4。
2.2.7 重复性试验 精密称取同一批配伍药材6份,按“2.2.2”项下进行操作,制成供试品溶液,测定没食子酸的含量 (mg/g),结果平均含量为32.477 mg/g,RSD为1.29%,表明重复性良好。
2.2.8 加样回收率试验 精密称取2.2.7项下已知含量的同一批次配伍药材2.5 g,置具塞锥形瓶中,精密加入1.048 mg/mL的没食子酸对照品溶液1.1 mL,按“2.2.2”项下供试品溶液制备项下方法制备供试液,在上述色谱条件下测定没食子酸含量,计算平均加样回收率和RSD。结果样品中没食子酸的平均回收率为98.11%,RSD为2.40%。结果见表5。

表4 稳定性试验考察结果

表5 加样回收率试验考察结果
2.3 总黄酮含量测定[11]
2.3.1 供试品溶液制备 精密称定配伍药材(吉祥草:余甘子=2∶1)粗粉0.5 g,精密加入60%乙醇50 mL,回流提取1.5 h,取出,放冷,用60%乙醇补足减失的重量,滤过,滤液作为供试品溶液,备用。
2.3.2 对照品溶液的制备 精密称取芦丁对照品5.12 mg,60%乙醇微热溶解并定容至25 mL,摇匀,得浓度为0.204 8 mg/mL的对照品溶液。
2.3.3 最大吸收波长的测定 准确吸取芦丁对照品溶液1.0 mL,置于具塞试管中,加入60%乙醇至5 mL,再加入5%亚硝酸钠0.3 mL摇匀,放置6 min;加入10%硝酸铝0.3 mL摇匀,放置6 min;加入1%NaOH溶液4 mL;加水0.4 mL,摇匀后放置15 min,在200~800 nm处进行最大吸光度的扫描。结果表明波长在508 nm处有最大吸光度。
2.3.4 线性关系考察 准确吸取芦丁对照品溶液0.0、1.0、2.0、3.0、4.0、5.0于6只具塞试管中,分别加入体积分数为60%乙醇溶液至5 mL;加入5%亚硝酸钠0.3 mL摇匀,放置6 min;加入10%硝酸铝0.3 mL摇匀,放置6 min;加入1% NaOH溶液4 mL;加入水0.4 mL,摇匀后放置15 min,以0.0 mL作为空白,于508 nm处测定各自吸光度,并以芦丁含量的浓度作为横坐标,以该浓度下所测定的吸光度作为纵坐标,绘制标准曲线并求出该标准曲线下对应的回归方程,回归方程和相关系数分别为:Y=10.493X-0.012,R2=0.9996,结果表明,芦丁在0.02~0.10 mg/mL范围内呈良好的线性关系。如图6所示。
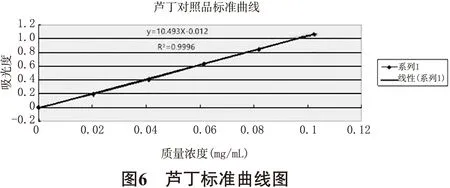
2.3.5 精密度试验 精密吸取芦丁对照品溶液2 mL,共6份,按“2.3.3”项下显色方法显色,于508 nm处测定吸光度,结果见表,计算吸光度RSD值为1.77%。结果表明,该方法精密度良好。
2.3.6 稳定性试验 精密吸取供试品溶液0.4 mL,按“2.3.3”项下显色方法显色,于508 nm处分别在0,15,30,45,60,90 min测定吸光度,结果见表7;计算吸光度RSD值为1.16%。结果表明,该方法表明样品溶液在90 min内基本保持稳定,所以,应该在样品显色后90 min内进行吸光度的测定。结果见表6。
2.3.7 重复性试验 精密称取2.3.6项下已知含量的同一批次配伍药材6份,按“2.3.1”项下供试品溶液的制备方法制备供试品溶液,取0.4 mL供试品溶液照“2.3.4”项下标准曲线的制备方法,自“加入60%乙醇溶液至5 mL”起,依法测定吸光度,计算含量,结果平均含量为8.75%,RSD为2.01%,表明重复性良好。
2.3.8 加样回收率试验 精密称取2.3.7项下已知含量的同一批配伍药材2.5 g,共6份,精密加入5 mL浓度为4.32 mg/mL的芦丁标准品溶液,摇匀,按供试品制备方法制备供试品溶液,取供试品溶液样品1 mL,照标准曲线的制备项下的方法,自“加入60%乙醇溶液至5 mL”起,依法测定吸光度。结果计算回收率为96.62%,RSD为1.48%。结果见表7。
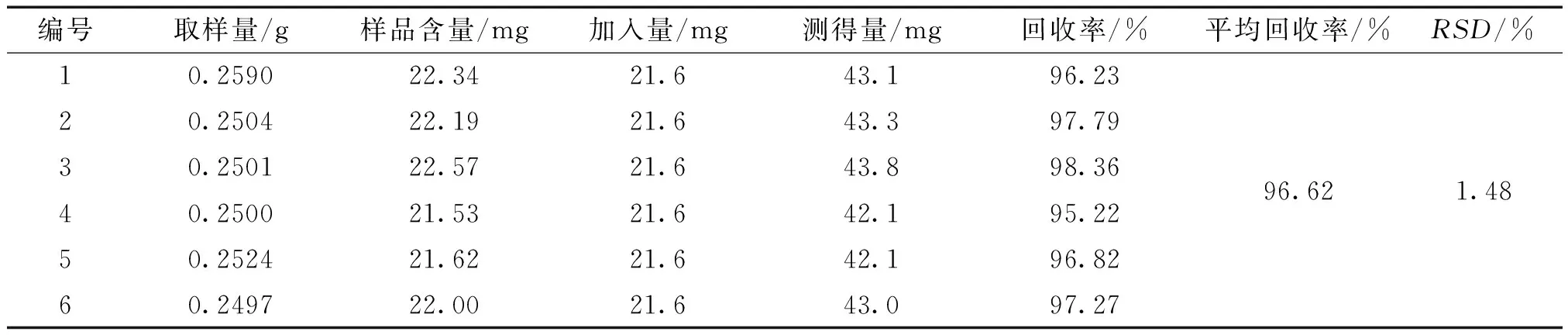
表7 加样回收率试验 (n=6)
2.4 供试品提取工艺优化
2.4.1 因素与水平设计 前期对吉祥草配伍余甘子不同提取物及有效部位组合药效学进行比较研究后,筛选出最佳提取方法为60%乙醇渗漉提取,为使有效部位中各有效成分最大程度的富集,以浸泡时间、60%乙醇用量、渗漉速度作为因素,每个因素设3个水平,最后以总皂苷含量、没食子酸含量、总黄酮含量总和以及得膏率为评价指标,采用L9(34)正交试验设计筛选最佳的有效部位提取方法,分析见表8。
2.4.2 正交试验及结果 称取吉祥草36g,余甘子18g,浸泡前先用60%乙醇54mL浸润30min。分别按表8条件进行试验,制得共9号干膏。吉祥草配伍余甘子渗漉提取工艺中各因素对结果影响的主次顺序为:C>B>A。具方差分析结果可知,各因素对实验结果没有显著的影响,综合考虑各方面因素,实验最终选择最佳的渗漉工艺为:A1B2C1即药材经过浸润后浸泡4 h,提取溶剂用量为15倍量,渗漉速度为1 mL/min。
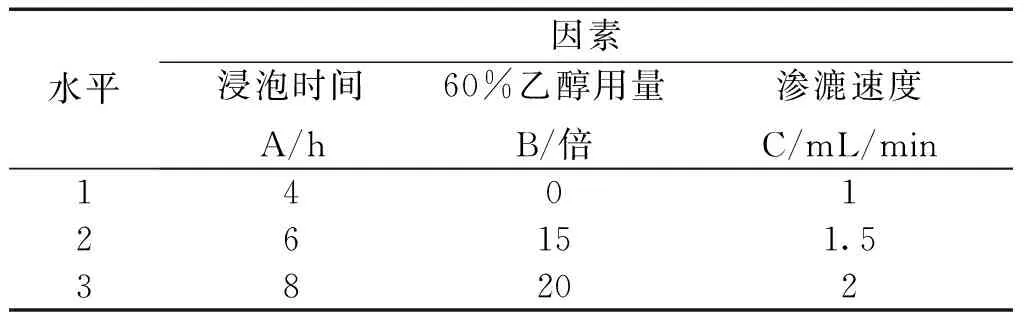
表8 因素水平表
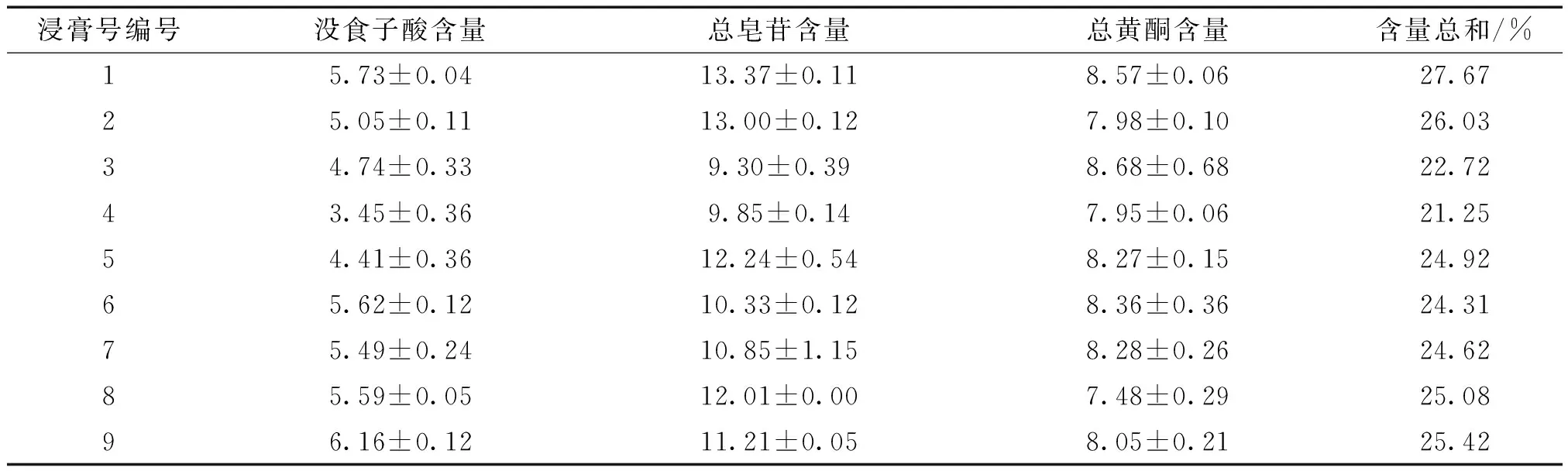
浸膏号编号没食子酸含量总皂苷含量总黄酮含量含量总和/%15.73±0.0413.37±0.118.57±0.0627.6725.05±0.1113.00±0.127.98±0.1026.0334.74±0.339.30±0.398.68±0.6822.7243.45±0.369.85±0.147.95±0.0621.2554.41±0.3612.24±0.548.27±0.1524.9265.62±0.1210.33±0.128.36±0.3624.3175.49±0.2410.85±1.158.28±0.2624.6285.59±0.0512.01±0.007.48±0.2925.0896.16±0.1211.21±0.058.05±0.2125.42
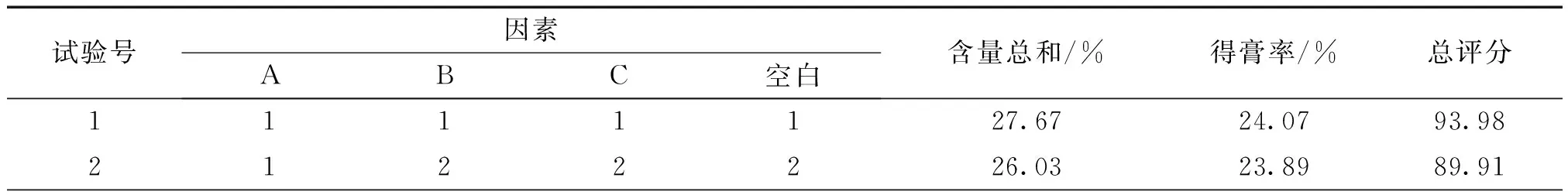
表10 正交试验设计及结果
续表
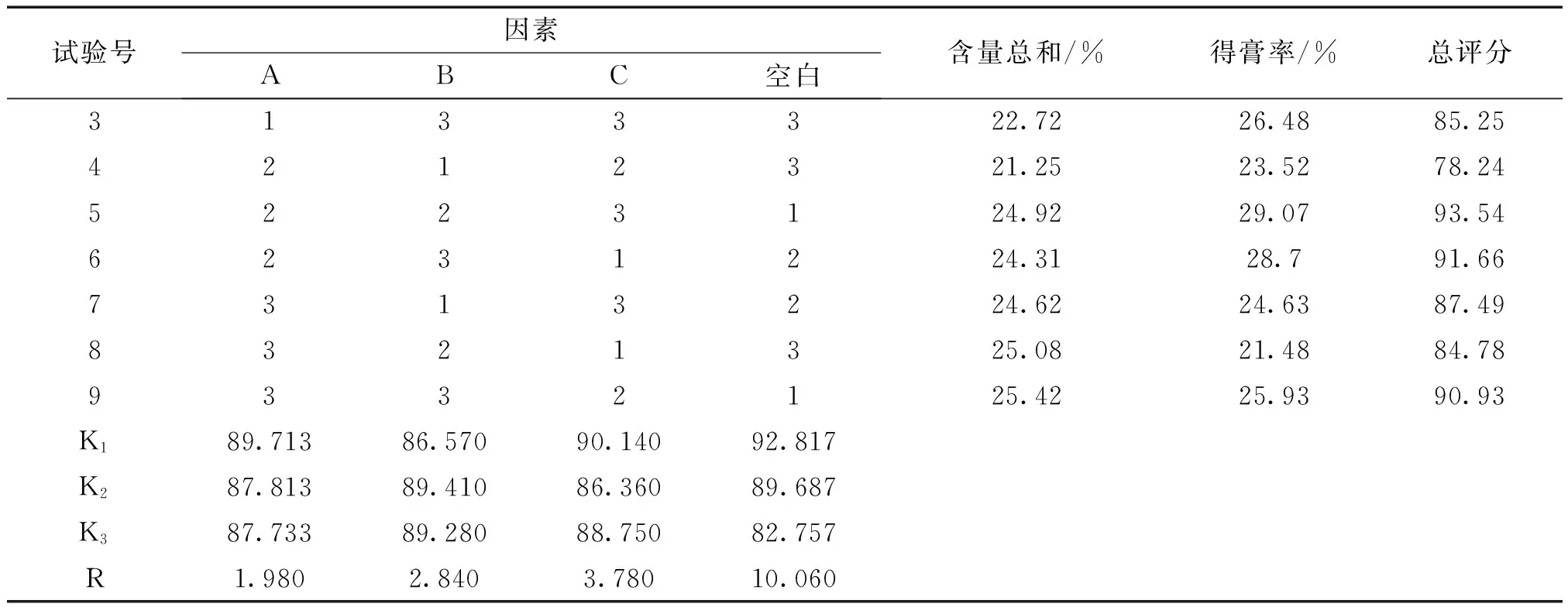
表10 正交试验设计及结果
总评分=(含量总和测定值/含量总和最大值)×65+(得膏率测定值/得膏率最大值)×35
2.4.3 验证实验 称取吉祥草36g,余甘子18g,3批,浸泡前先用60%乙醇54mL浸润30min后浸泡4h,用提取溶剂用为15倍量渗漉,渗漉速度为1mL/min,渗滤液减压干燥得干膏,按上述建立含量测定方法进行含量测定,表明含量测定符合优选出的工艺,工艺稳定,合理可行。结果见表11。
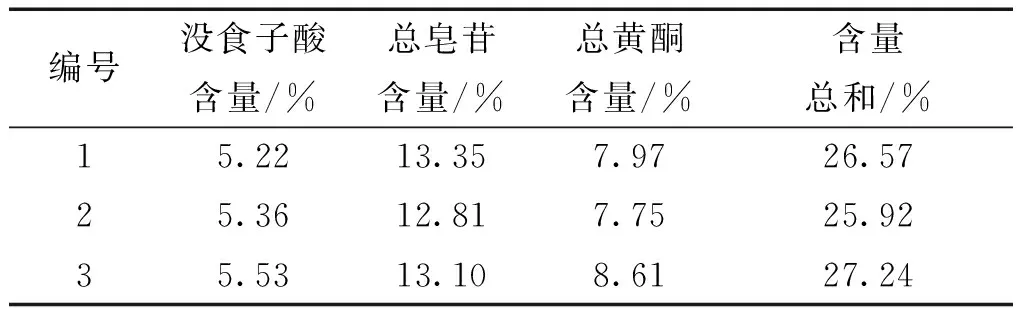
表11 验证实验结果
3 讨论
吉祥草与余甘子(2∶1)配伍,分别采用不同的提取方法,所得浸膏配成药液后作用于非小细胞肺癌实验动物模型,结果对肿瘤模型小鼠生活质量有不同程度的改善,其中60%乙醇渗漉提取法优于其他方法。研究显示,60%乙醇渗漉提取部位能够较大程度的富集黄酮、皂苷等主要抗肿瘤成分,故采用60%乙醇渗漉法提取。为使最佳提取部位中各有效成分最大程度富集,以浸泡时间、60%乙醇用量以及渗漉速度作为因素,并以没食子酸含量、总黄酮含量、总皂苷含量总和以及得膏率作为评价指标,采用L9(34)正交试验设计优化60%乙醇渗漉提取工艺。最终选择最佳条件是药材浸润后浸泡4h,提取溶剂用量为15倍量,渗漉速度为1mL/min。
[1]邱德文,杜江.中华本草苗药卷[M].贵阳:贵州科技出版社,2005.
[2]黄诗娅,李江,马国新.用于COPD的吉祥草雾化吸入剂制备及其质量控制研究[J].亚太传统医药,2014,10(22):19-20.
[3]刘海,杨建琼,熊亮,等.吉祥草提取物对人结肠癌HT-29细胞体外抑制作用的研究[J].时珍国医国药,2013,24(5):1103-1105.
[4]白彩虹,邹坤,贺海波.吉祥草乙酸乙酯部位对人宫颈癌Caski细胞抑制作用及COX-2基因表达与BCl-2蛋白家族关系的研究[J].中药药理与临床,2013,29(6):45-49.
[5]付雪娇,邹坤,王桂萍.吉祥草乙酸乙酯提取物抗炎作用及机制研究[J].时珍国医国药,2013,24(4):822-825.
[6]邱德文,杜江.贵州十大苗药研究[M].北京:中医古籍出版社,2008.
[7]蔡英卿.我国余甘子种质资源与开发利用研究的进展[J].福建果树,2000,13(1):18-20.
[8]周婵媛,陈华国,周欣,等. 紫外分光光度法测定吉祥草中的总皂苷[J]. 华西药学杂志,2010,25(3):344-346.
[9]顿珠,刘青,索朗其美,等. HPLC法测定余甘子膏中没食子酸[J].中草药,2010,41(9):1477-1478.
[10]邹丽,高向军,包小红,等. HPLC法测定余甘子中没食子酸的含量[J].食品与药品,2009,11(7):49-51.
[11]李舒,钟振国,赖进科. 余甘子叶提取物总黄酮的含量测定[J].辽宁中医药大学学报,2011,13(7):109-110.
