超高效液相色谱-四极杆/静电场轨道阱高分辨质谱快速筛查水产及水产加工品中24种镇静剂类药物
2018-03-05杨璐齐张鹏云罗阳丹张朋杰高永清
李 蓉, 杨璐齐, 张鹏云, 罗阳丹, 张朋杰, 高永清
(1. 中山出入境检验检疫局检验检疫技术中心, 广东 中山 528400; 2. 广东药科大学食品科学学院, 广东 中山 528458)
镇静剂又称为运动抑制剂,是指对中枢神经系统有抑制作用,可起到减轻或消除动物在高密度饲养或长途运输、转群、高温等应激情况下的狂躁不安,使其恢复安静的一类药物[1]。镇静剂残留在动物组织中可引发食品安全问题[2]。正常人摄入含有镇静剂的食品会造成肝脏负担加重,头脑长期昏沉,记忆受损,同时运动神经和肌肉功能受到抑制[3]。我国农业部公告(第235号)规定安眠酮为禁止使用的药物,在动物性食品中不得检出;氯丙嗪、地西泮、塞拉嗪为允许做治疗使用但不得在动物性食品中检出的药物[4]。镇静剂被卫生部列入食品中可能违法添加的非食用物质名单[5]。国际食品法典委员会(CAC)、欧盟、澳大利亚等国家和组织均对镇静剂的检出和最高限量值做出了相关规定[1]。 国内外镇静剂的检测方法有酶联免疫法(ELISA)[6,7]、气相色谱-质谱联用法(GC-MS)[8,9]、液相色谱法(LC)[3,10]、液相色谱-串联质谱法(LC-MS/MS)[1,2,11-14]、液相色谱-四极杆飞行时间质谱法(LC-Q-TOF MS)[15-18]等。ELISA稳定性和重复性较差,假阳性率较高;LC灵敏度较低,以保留时间定性,不能用于确证;GC-MS的灵敏度较好,但前处理时需结合固相萃取柱净化,操作步骤繁琐。LC-MS/MS具备较高的灵敏度和特异性,是目前国内外检测镇静剂时常用的一种定性定量方法。近年来高分辨质谱开始得到有效应用,如用LC-Q-TOF MS测定饲料[15]、牛肉[16]、牛奶[17]和乳制品[18]中的镇静剂。超高效液相色谱-四极杆/静电场轨道阱高分辨质谱(UPLC-Q-Orbitrap HRMS)具有高灵敏度、高选择性和高质量精度的优点,可用于农药残留[19]、降糖药物[20]、多肽类药物[21]、真菌毒素[22]等的测定,尚未见用其分析水产类样品中的镇静剂。
本文建立了UPLC-Q-Orbitrap HRMS同时测定水产及水产加工制品中24种镇静剂的分析方法。与LC-MS/MS相比,本方法的检出限更低,线性范围更宽,分析精度和准确度更高,且实验操作步骤简单,适用于大批量水产及水产加工品中24种镇静剂类药物的筛查。
1 实验部分
1.1 仪器、试剂与材料
超高效液相色谱-四极杆/静电场轨道阱高分辨质谱仪(UltiMate 3000 Q Exacitve UPLC-HRMS,配可加热电喷雾离子(HESI)源,TraceFinder 3.3 EFS数据处理系统,美国Thermo Fisher公司); ACQUITY UPLC®BEH C18色谱柱(100 mm×2.1 mm, 1.7 μm,美国Waters公司); GM 200捣磨仪(德国Retsch公司); CP2250天平(德国Sartorius公司); T18涡旋混合器(德国IKA公司); MMV-1000W超声振荡器(日本Eyela公司); Synocre平行定量浓缩仪(瑞士Buchi公司); Milli-Q纯水机(美国Millipore公司); 2-16K台式离心机(德国Sartorius-Sigma公司); 0.22 μm有机滤膜(上海安谱公司)。
乙腈、正己烷、甲醇、叔丁基甲醚和乙酸乙酯(HPLC级,美国Merck公司);氯化钠、无水硫酸镁和无水硫酸钠(分析纯,广州化学试剂厂)。
艾司唑仑(estazolam)、咪哒唑仑(midazolam)、三唑仑(triazolam)、阿普唑仑(alprazolam)、硝西泮(nitrazepam)、氯硝西泮(clonazepam)、氟硝西泮(flunitrazepam)、奥沙西泮(oxazepam)、去甲西泮(nordazepam)、安眠酮(methaqualone)和地西泮(diazepam)均为0.1 g/L的标准溶液,溶剂为甲醇;劳拉西泮(lorazepam)为0.1 g/L的标准溶液,溶剂为乙腈,以上12种标准溶液均购自德国LGC Standards公司。丙酰二甲基丙吩噻嗪(propionylpromazine,纯度97.8%)、阿扎哌隆(azaperone,纯度98.5%)、乙酰丙嗪(acepromazine,纯度98.0%)、氯丙嗪(chlorpromazine,纯度99.0%)、阿扎哌醇(azaperol,纯度98.0%)、异丙嗪(promethazine,纯度99.0%)、咔唑心安(carazolol,纯度99.0%)、塞拉嗪(xylazine,纯度99.0%)8种标准品均购自德国Dr. Ehrenstorfer公司。氟奋乃静(fluphenazine,纯度98.8%)标准品购自北京曼哈格生物科技。氟哌啶醇(haloperidol,纯度98%)、奋乃静(prochlorperazine,纯度96%)、氟哌利多(droperidol,纯度98%)3种标准品均购自加拿大Toronto Research Chemicals公司。
实验用样品均购自当地市场。
1.2 标准溶液的配制
分别取适量上述标准品,地西泮、艾司唑仑、咪哒唑仑、三唑仑、阿普唑仑、氯硝西泮、氟硝西泮、奥沙西泮、去甲西泮和安眠酮以甲醇为溶剂,劳拉西泮、硝西泮以乙腈为溶剂,配制成质量浓度为10 mg/L的单标准储备液;氟哌啶醇、塞拉嗪、咔唑心安以无水乙醇为溶剂,氟哌利多、异丙嗪、氯丙嗪、阿扎哌醇、阿扎哌隆、氟奋乃静、乙酰丙嗪、奋乃近、丙酰丙嗪以甲醇为溶剂,配制成质量浓度为100 mg/L的单标准储备液。
将24种镇静剂单标准储备液用甲醇配制成质量浓度为1 mg/L的混合标准溶液,于4 ℃冰箱中避光保存;用50%(v/v)甲醇水溶液稀释成所需浓度的混合标准工作溶液,现用现配。
1.3 样品前处理
准确称取均质后的试样(2.00±0.01) g,置于50 mL聚丙烯离心管中,加入4 g氯化钠,涡旋30 s混匀,加入10 mL乙腈,均质1 min,超声10 min,以4 000 r/min的转速离心5 min,将上清液转移至另一50 mL聚丙烯离心管中。用10 mL乙腈清洗均质机的刀头,清洗液置于残留有残渣的离心管中,振摇10 min,以4 000 r/min的转速离心5 min,合并上清液,涡旋混匀,移取10 mL上清液,用平行定量浓缩仪浓缩至近干,用1 mL 50%(v/v)甲醇水溶液定容,加入6 mL乙腈饱和的正己烷溶液,以2 000 r/min的转速涡旋30 s,超声5 min,静置15 min,取1.2 mL下层溶液转移至2 mL高速离心管中,于-20 ℃冰箱中放置30 min然后于4 ℃以14 500 r/min的转速离心15 min。取下层溶液过0.22 μm有机滤膜,待上机分析。
1.4 混合基质标准工作液的配制
实际样品检测时,取同类阴性样品,按1.3节操作浓缩至近干后,加入1.0 mL质量浓度为100、50、25、5、2.5、1.0、0.5和0.25 μg/L的混合标准溶液复溶,涡旋混匀,净化,过0.22 μm有机滤膜,滤液待上机分析。
1.5 分析条件
1.5.1 色谱条件
色谱柱:Waters ACQUITY UPLC®BEH C18柱(100 mm×2.1 mm, 1.7 μm);柱温:30 ℃;流动相:A相为0.1%(v/v)甲酸水溶液,B相为含0.1%(v/v)甲酸的乙腈溶液;流速:0.3 mL/min。梯度洗脱条件:0~15.0 min, 95%A; 15.0~17.0 min, 5%A; 17.0~17.1 min, 95%A; 17.1~20.0 min, 95%A。进样量:10 μL。
1.5.2 质谱条件
离子源:HESI源,正离子扫描模式;采集方式:全扫描/数据依赖二级扫描(Full MS/dd-MS2), TopN=1(前1强);一级扫描分辨率:70 000;二级扫描分辨率:17 500;归一化碰撞能:20%、40%、60%;喷雾电压:3 200 V;离子传输管温度:325 ℃;加热器温度:350 ℃;鞘气(N2)流速:40 Arb;辅助气(N2)流速:10 Arb。动态排除:6 s;顶点触发:2~6 s。24种镇静剂药物的其他质谱参数见表1。

表 1 24种镇静剂类药物的分子式、保留时间、母离子和碎片离子Table 1 Molecular formulas, retention times (RTs), precursor ions and fragment ions of the 24 tranquillizer drugs
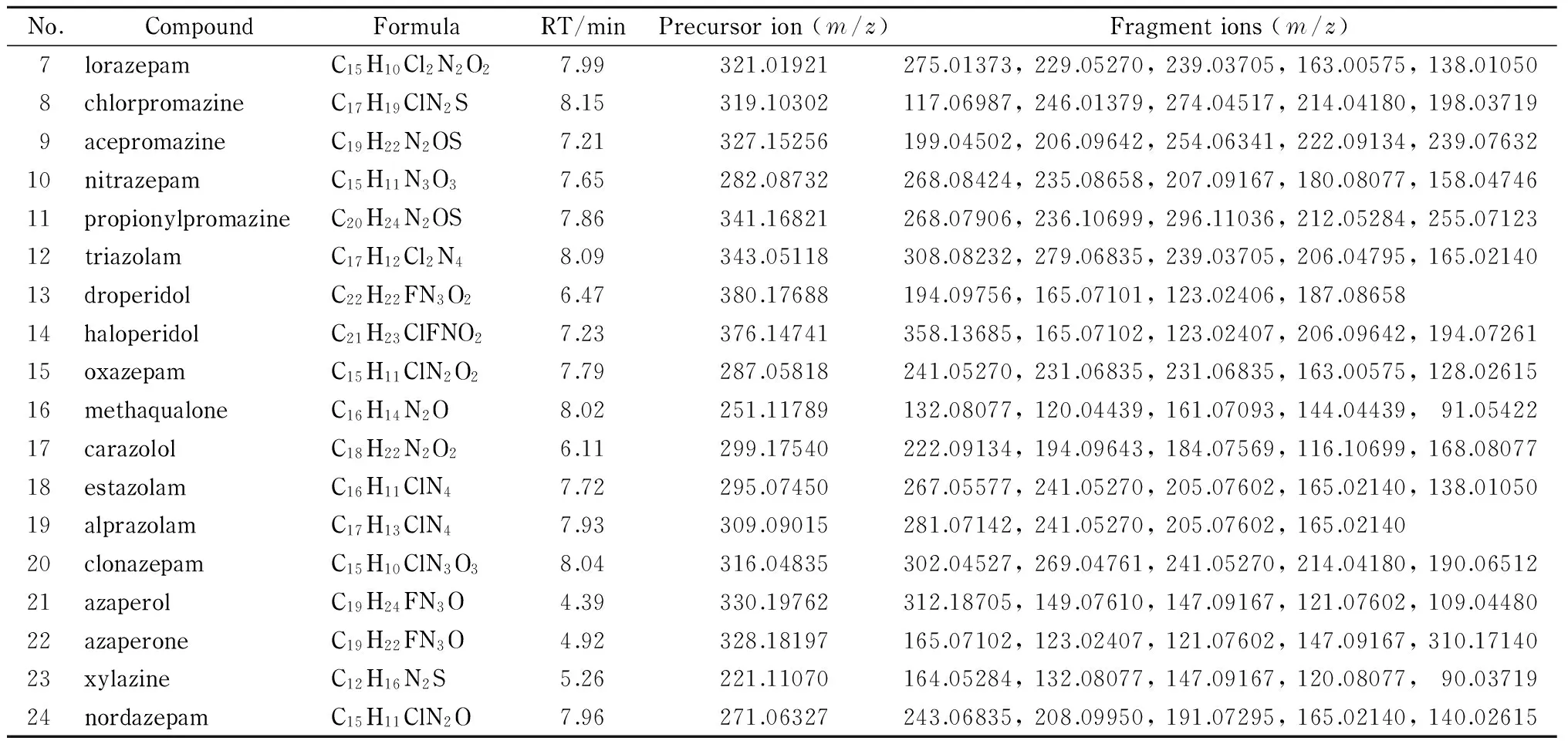
表 1 (续)Table 1 (Continued)
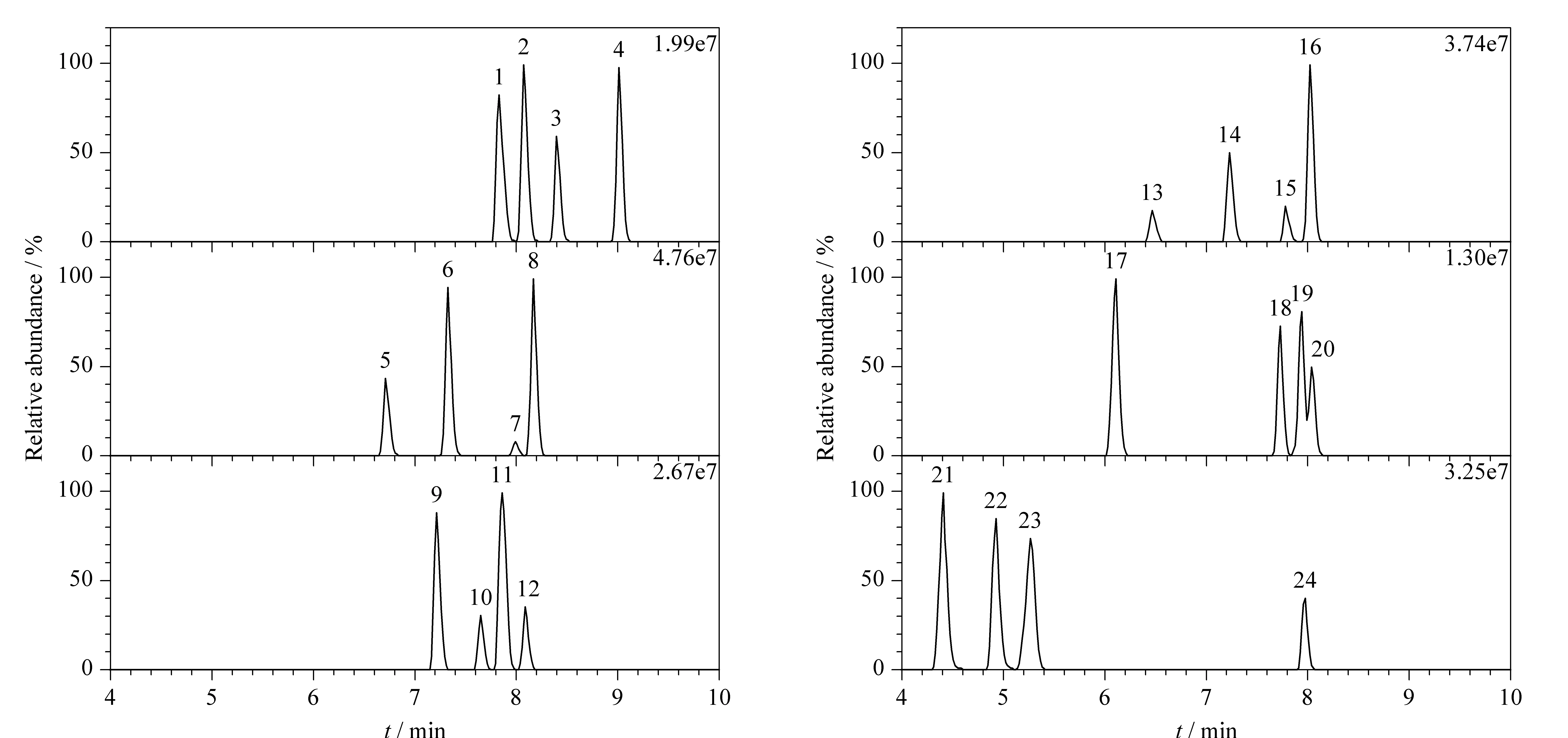
图 1 加标鲩鱼基质溶液中24种镇静剂(10 μg/L)的提取离子流色谱图Fig. 1 Extracted ion chromatograms of the 24 tranquilizer drugs (10 μg/L) spiked in the grass carp matrix solutionNos. 1-24 were the same as that in Table 1.
2 结果与讨论
2.1 色谱条件的优化
实验对比了Waters ACQUITY UPLC®BEH C18色谱柱(100 mm×2.1 mm, 1.7 μm)和Thermo Fisher Accucore RP-MS色谱柱(100 mm×2.1 mm, 2.6 μm)的分离效果。结果发现,Waters ACQUITY UPLC®BEH C18色谱柱的粒径更小,柱效较高,分离效果更好。流动相A选择0.1%(v/v)甲酸水溶液,流动相B选择含0.1%(v/v)甲酸的乙腈溶液[12],采用梯度洗脱程序。加标鲩鱼基质溶液中24种镇静剂(10 μg/L)的提取离子流色谱图见图1。
2.2 质谱条件的确定
使用一级质谱全扫描加数据依赖二级质谱扫描方式对选定的质量数(m/z100~1 000)做一级质谱全扫描,发现母离子,其强度达到设定阈值(1×106)时,自动触发二级质谱扫描。在得到母离子精确质量数的同时又得到了二级质谱的全扫描信息。采用20%、40%、60% 3种不同归一化碰撞能对化合物进行碎裂,得到一张碎片离子信息丰富的加合图。进一步调整动态排除、顶点触发和TopN等参数来获取较好的碎片离子信息。动态排除时间对比了6、8和10 s,结果表明,6 s时24种目标物的峰形较好,当顶点触发为2~6 s、TopN=1时可获得满意的二级碎片离子信息和二级质谱图。
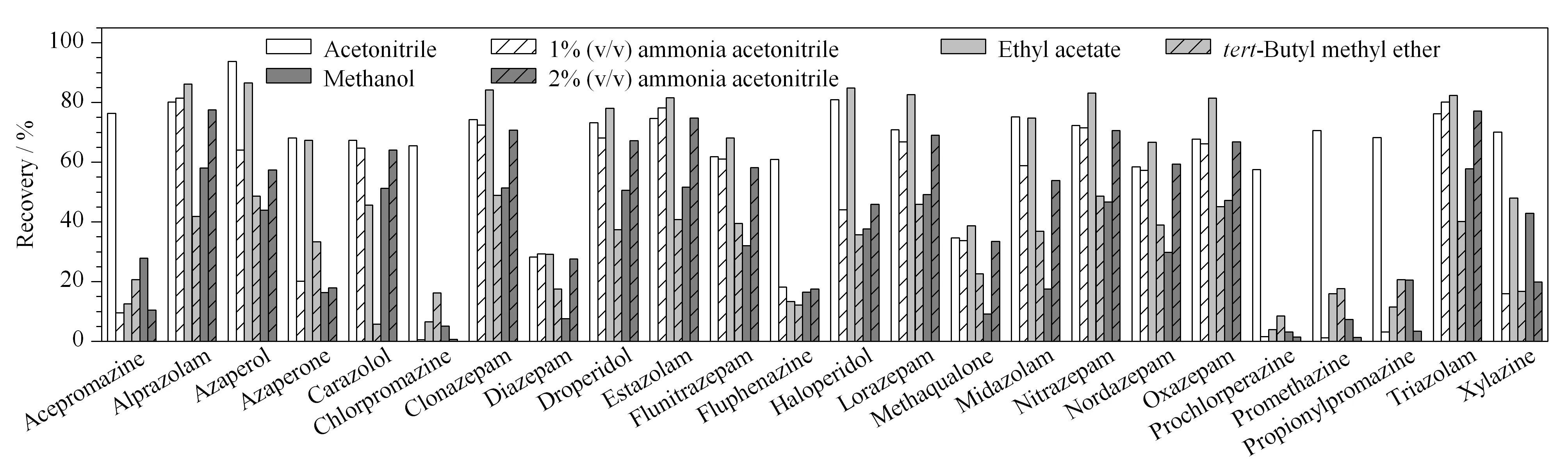
图 2 不同提取溶剂对24种镇静剂类药物回收率的影响Fig. 2 Effects of the different extraction solvents on the recoveries of the 24 tranquillizer drugs
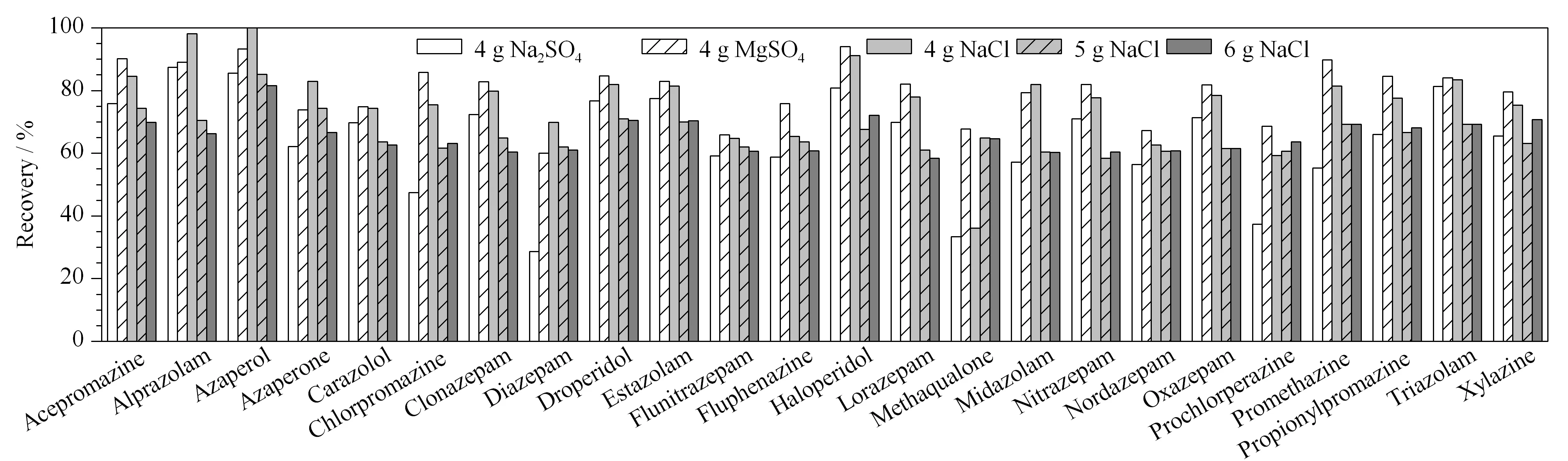
图 3 不同盐析剂对24种镇静剂类药物回收率的影响Fig. 3 Effects of different salting-out agents on the recoveries of the 24 tranquillizer drugs
用50%(v/v)甲醇水溶液配制质量浓度为100 μg/L的混合标准溶液,在Full MS/dd-MS2(TopN=1)方式下进行扫描,确定目标物为[M+H]+的准分子离子峰。依据目标物的分子式和碎片离子信息拟合出理论精确质量数。24种目标化合物的母离子、碎片离子等信息见表1。
2.3 样品前处理条件的优化
2.3.1 样品提取溶剂的选择
本实验将10 mL的乙腈、含1%(v/v)氨水的乙腈溶液、乙酸乙酯、叔丁基甲醚、甲醇、含2%(v/v)氨水的乙腈溶液作为样品提取溶剂,对比了24种镇静剂的提取效率(见图2)。结果显示,使用乙腈提取时各目标物均可获得较好的回收率,为进一步提高提取效率,实验发现使用10 mL乙腈提取两次的效果更好。
2.3.2 盐析剂及用量的选择
前处理过程中加入盐析剂有利于有机相与水相的分层,防止样品中的水分及杂质进入提取液,从而提高目标物的回收率。实验对比了无水硫酸镁、无水硫酸钠和氯化钠(均4 g)等盐析剂对目标物回收率的影响(见图3)。结果发现,加入无水硫酸钠时,地西泮、奋乃静、氯丙嗪、安眠酮等物质的回收率明显偏低;加入氯化钠或无水硫酸镁时,24种目标物的回收率均较高且差别不大,本实验选择更常用的氯化钠作为盐析剂。进一步对比添加4、5和6 g氯化钠时的效果,结果显示,各目标物的添加回收率无显著性差异(见图3),因此最终选择4 g氯化钠作为盐析剂。
2.3.3 复溶液的选择
有文献报道可使用50%(v/v)甲醇水溶液[12]和30%(v/v)乙腈水溶液[19]作为浓缩近干后的复溶液,实验首先对比了30%(v/v)甲醇水溶液和30%(v/v)乙腈水溶液对目标物回收率的影响,结果显示,使用30%(v/v)甲醇水溶液的回收率较高。因此,进一步对比了体积分数为30%、50%和60%的甲醇水溶液对目标物回收率的影响,发现使用50%(v/v)的甲醇水溶液时,各目标物均可获得满意的回收率(见图4)。
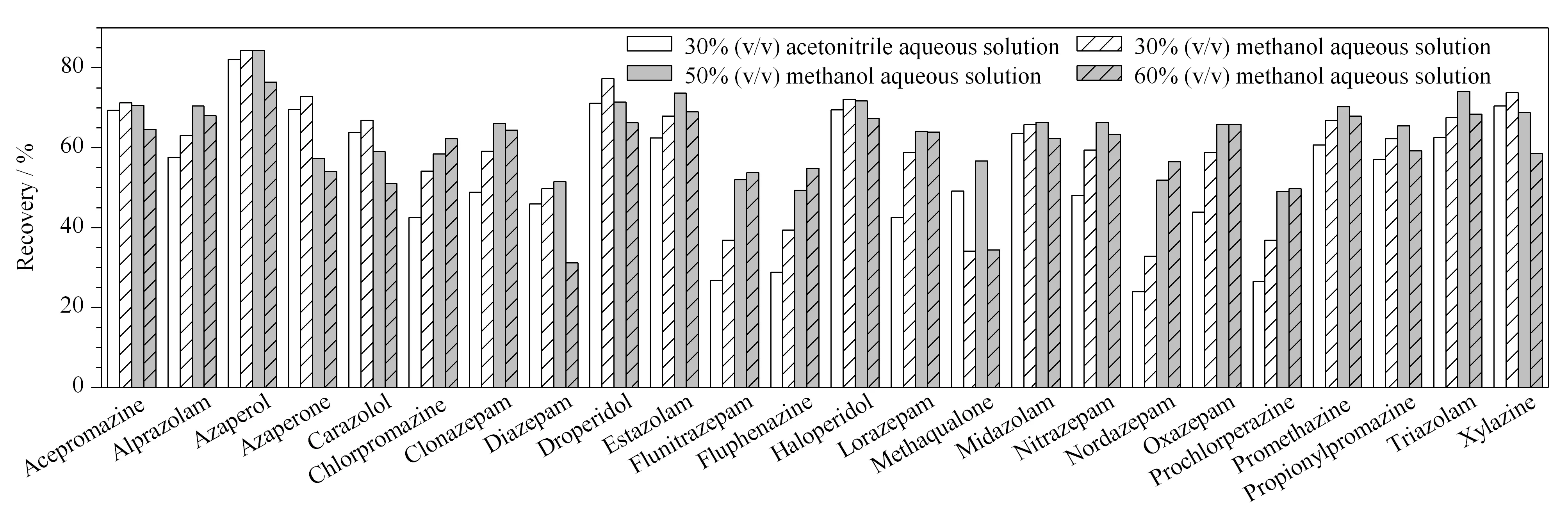
图 4 不同复溶液对24种镇静剂类药物回收率的影响Fig. 4 Effects of the different reconstitution solutions on the recoveries of the 24 tranquillizer drugs
2.4 线性方程和定量限
逐级稀释配制不同质量浓度(300、200、100、50、25、10、5、2.5、1.0、0.5、0.25、0.125、0.062 5和0.031 25 μg/L)的混合标准工作溶液。在1.5节的分析条件下,以各化合物的质量浓度(x, μg/L)为横坐标、对应的峰面积(y)为纵坐标建立标准曲线,得到各化合物的线性范围、线性方程及其对应的决定系数(r2)。结果显示,24种镇静剂类药物在各自的范围内线性关系良好,r2≥0.996 8(见表2)。
本文对鲜鲩鱼、墨鱼丸、鲮鱼罐头、盐制鱿鱼干、冰冻南美对虾、面线鱼干进行了基质效应(matrix effect, ME)的评估。计算公式为ME=(A-B)/B×100%。其中,A和B分别为目标物(10 μg/L)在基质匹配标准溶液和50%(v/v)甲醇水溶液中的仪器响应值。基质效应为负值表示基质抑制效应,正值表示基质增强效应[23],具体结果见表2。在6种基质中,24种镇静剂类药物中有41.7%属于弱基质效应(-10%≤ME≤10%), 43.8%属于强基质增强效应(ME>10%), 14.6%属于强基质抑制效应(ME<-10%)。因此,为降低基质效应,更准确地定量,本文采用基质标准工作液进行定量。
高分辨质谱通过提取目标物的精确质量数来进行定量,无法获取空白样品的测量值,不能按GB/T 27404-2008《实验室质量控制规范食品理化检测》标准通过计算得到测定低限(CL)。同时,因大部分目标化合物色谱图的基线噪声极低,也不适宜用信噪比来确定方法的定量限(LOQ)[24]。本方法利用空白基质做加标回收试验,将满足回收率要求的最低加标浓度点作为方法的定量限。结果显示,方法的定量限范围为0.1~1.0 μg/kg(见表2)。
2.5 回收率与精密度
为验证方法的准确度,分类选取有代表性的空白水产及水产加工品基质(鲜鲩鱼、墨鱼丸、鲮鱼罐头、盐制鱿鱼干、冰冻南美对虾、面线鱼干)进行3个水平的加标回收试验,每个添加水平重复测定6次,目标化合物的回收率为58.9%~122.9%, RSD为0.2%~16.4%(见表S1,详见http://www.chrom-China.com/UserFiles/File/1710002-SI.pdf)。
2.6 实际样品检测
用建立的方法对购买的70份水产及水产加工品进行筛查,其中50%为鲜活水产品,包括桂鱼、鲫鱼、青口、沙虾、白贝、北极贝、细足趸、丁桂鱼、瓜子斑、明虾、老虎仔鱼、白贝、罗氏沼虾、全壳贻贝、鲮鱼、濑尿虾、罗非鱼、面线鱼、鲩鱼、肉蟹、大鱼头和兰尾对虾等35个样品。结果表明,在濑尿虾中检出异丙嗪,含量为0.977 μg/kg;在某鱼罐头样品中检出安眠酮,含量为1.039 μg/kg(见图5)。安眠酮易通过胎盘屏障导致畸胎,通过乳汁引起新生儿昏睡,通过血脑屏障导致中枢神经抑制[25],长期摄入残留安眠酮的动物产品,人体可产生耐药性[26]。在农业部公告第193号中,安眠酮被明确列入《禁用清单》。水产加工品中检出镇静剂的原因可能是:一、环境因素,如养殖水被污染;二、养殖户误用含有镇静剂的饲料;三、在养殖和运输过程中的人为添加。

表 2 24种镇静剂类药物的线性范围、线性方程、决定系数、基质效应和定量限(n=6)Table 2 Linear ranges, linear equations, determination coefficients (r2), matrix effects (MEs) and LOQs of the 24 tranquillizer drugs (n=6)

表 2 (续)Table 2 (Continued)
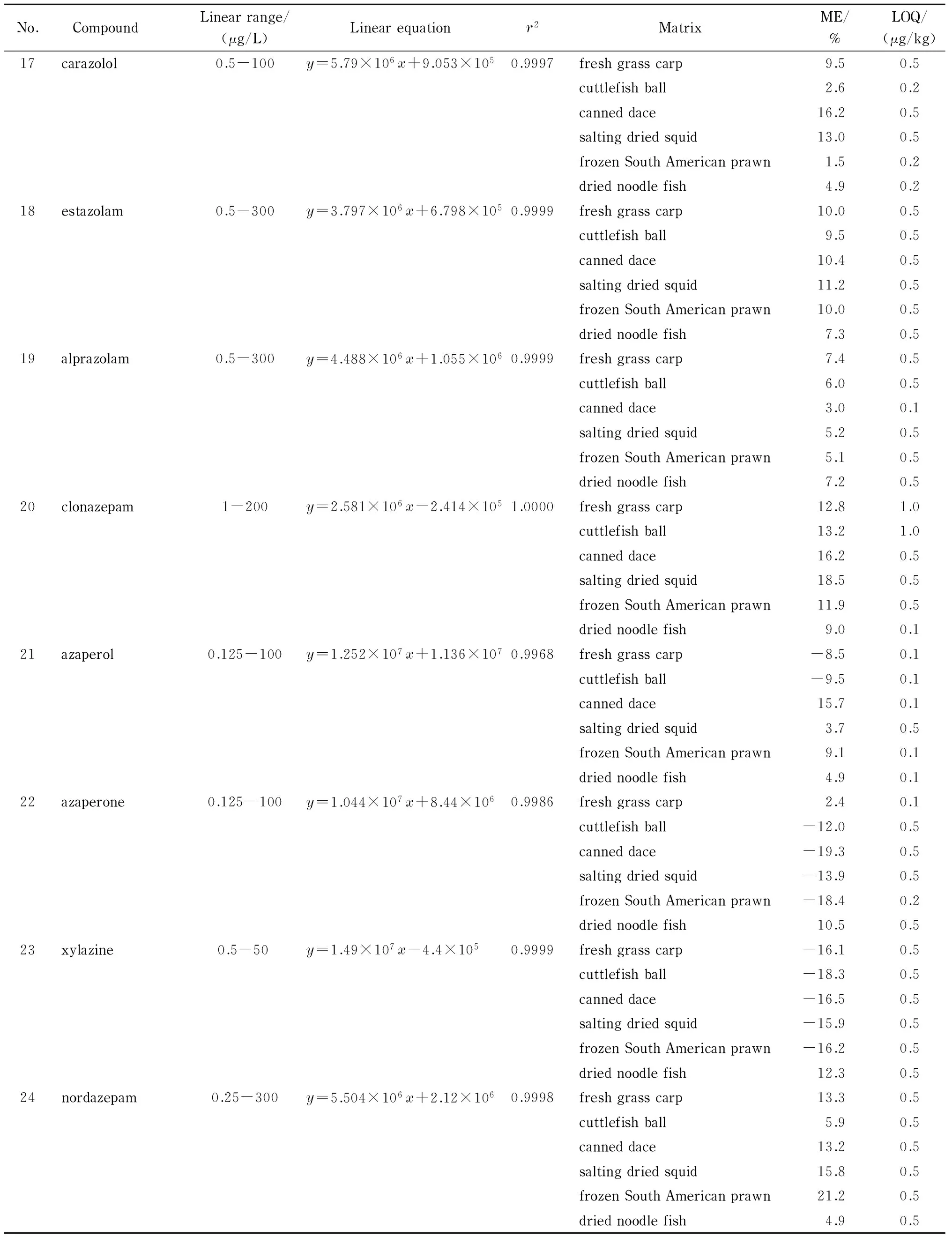
表 2 (续)Table 2 (Continued)
y: peak area;x: mass concentration, μg/L.

图 5 阳性样品中安眠酮的提取离子流色谱图和二级质谱图Fig. 5 Extracted ion chromatograms and MS2 spectrum of methaqualone in the positive sample a. extracted precursor ion chromatogram in Full MS mode; b. extracted precursor ion chromatogram in Full MS2 mode; c. MS2 spectrum.
3 结论
本研究通过对样品前处理条件及仪器参数的优化,建立了UPLC-Q-Orbitrap HRMS测定水产及水产加工品中24种镇静剂类药物的分析方法。该方法操作简单,灵敏度高,准确可靠,适用于日常对大批量水产及水产加工品中多种镇静剂类药物的快速筛查及确证。
[1] Zhang S, Zhou S, Chen D W, et al. Journal of Instrumental Analysis, 2014, 33(11): 1213
张烁, 周爽, 陈达炜, 等. 分析测试学报, 2014, 33(11): 1213
[2] Wei J M, Luo Y Z, Zhang L, et al. J Sci Food Agric, 2015, 95(3): 598
[3] Wei J M, Luo Y Z, Bai Y X. Science and Technology of Food Industry, 2014, 35(10): 95
魏晋梅, 罗玉柱, 白云旭. 食品工业科技, 2014, 35(10): 95
[4] Ministry of Agriculture. No. 235 Bulletin of the Ministry of Agriculture of the People’s Republic of China. (2002-12-24) [2017-10-08]. http://yz.hz-agri.gov.cn/uploadFiles/2005-10/1130221564406.doc
农业部. 中华人民共和国农业部公告第235号. (2002-12-24) [2017-10-08]. http://yz.hz-agri.gov.cn/uploadFiles/2005-10/1130221564406.doc
[5] National Health and Family Planning Commission of the People’s Republic of China. The List of Possible Illegally Added Non-edible Substances and Easy-to-Abuse Food Additives in Food (1-5 Batches Summary). (2011-04-22) [2017-10-08]. http://www.nhfpc.gov.cn/sps/s7892/201406/38e5c8a53615486888d93ed05ac9731a.shtml
中华人民共和国国家卫生和计划生育委员会. 食品中可能违法添加的非食用物质和易滥用的食品添加剂名单(第1-5批汇总). (2011-04-22) [2017-10-08]. http://www.nhfpc.gov.cn/sps/s7892/201406/38e5c8a53615486888d93ed05ac9731a.shtml
[6] Peng C F, Duan X H, Song S S, et al. Int J Mol Sci, 2013, 14(10): 19474
[7] Wang Y Y, Jia T. Feed Research, 2008(11): 50
王有月, 贾涛. 饲料研究, 2008(11): 50
[8] Tan G L, Zhao T Z, Wang W L, et al. Modern Food Science and Technology, 2014, 30(2): 274
谭贵良, 赵天珍, 王文林, 等. 现代食品科技, 2014, 30(2): 274
[9] GB 29697-2013
[10] Uddin M N, Samanidou V F, Papadoyannis I N. J Sep Sci, 2008, 31(21): 3704
[11] Sun L, Zhang L, Xu Q, et al. Chinese Journal of Chromatography, 2010, 28(1): 38
孙雷, 张骊, 徐倩, 等. 色谱, 2010, 28(1): 38
[12] Doran G S, Bradbury L A. J Chromatogr B, 2015, 997: 81
[13] Zou Y, Shao L Z, Chen S M, et al. Chinese Journal of Chromatography, 2017, 35(8): 801
邹游, 邵琳智, 陈思敏, 等. 色谱, 2017, 35(8): 801
[14] De Oliveira L G, Barreto F, Hoff R, et al. Food Addit Contam, 2017, 34(1): 32
[15] Wu N P, Ban F G, Peng L, et al. Journal of Chinese Mass Spectrometry Society, 2012, 33(2): 94
吴宁鹏, 班付国, 彭丽, 等. 质谱学报, 2012, 33(2): 94
[16] Meng L, Wu N P, Hu X J, et al. Chinese Journal of Veterinary Drug, 2013, 47(8): 36
孟蕾, 吴宁鹏, 胡兴娟, 等. 中国兽药杂志, 2013, 47(8): 36
[17] Zhang Y Q, Li X, Liu X M, et al. J Dairy Sci, 2015, 98(12): 8433
[18] Yan L J, Zhang J, Pan C S, et al. Chinese Journal of Analytical Chemistry, 2013, 41(1): 31
严丽娟, 张洁, 潘晨松, 等. 分析化学, 2013, 41(1): 31
[19] Yu W, Liu J Y, Wang W L, et al. Environmental Chemistry, 2014, 33(12): 2225
余巍, 刘军义, 汪文龙, 等. 环境化学, 2014, 33(12): 2225
[20] Du Y S, Li Q, Wu C M, et al. Chinese Journal of Chromatography, 2015, 33(4): 371
杜彦山, 李强, 吴春敏, 等. 色谱, 2015, 33(4): 371
[21] Zhu W X, Yang J Z, Li S, et al. Chinese Journal of Chromatography, 2017, 35(2): 156
祝伟霞, 杨冀州, 李睢, 等. 色谱, 2017, 35(2): 156
[22] Li R, He C M, Yang L Q, et al. Chinese Journal of Chromatography, 2017, 35(8): 808
李蓉, 何春梅, 杨璐齐, 等. 色谱, 2017, 35(8): 808
[23] Zhang Z Y, Zhao L W, Cao W R, et al. Food Science, 2016, 37(16): 242
张中印, 赵柳微, 曹葳蕤, 等. 食品科学, 2016, 37(16): 242
[24] Mu H T B E, Yan H, Xu S, et al. Chinese Journal of Chromatography, 2015, 33(11): 1199
木合他拜尔, 严华, 徐姗, 等. 色谱, 2015, 33(11): 1199
[25] Sun G Q, Zhao H L, Rong J Z. Chinese Journal of Hospital Pharmacy, 1984, 4(12): 18
孙国清, 赵汉林, 戎建中. 中国医院药学杂志, 1984, 4(12): 18
[26] Deng Y B, Duan S H. Hunan Feed, 2003(3): 21
邓云波, 段苏华. 湖南饲料, 2003(3): 21
