QuEChERS-超高效液相色谱-四极杆/静电场轨道阱高分辨质谱快速测定水产品中25种药物残留
2018-03-05张宪臣张朋杰华洪波杨璐齐卢俊文容裕棠
张宪臣, 李 蓉, 张朋杰, 吴 霞,华洪波, 杨璐齐, 卢俊文, 容裕棠
(1. 中山出入境检验检疫局检验检疫技术中心, 广东 中山 528403; 2. 安捷伦科技(中国)有限公司, 广东 广州 510000; 3. 中山市卓雅外语学校, 广东 中山 528401)
随着现代养殖业集约化和规模化,水产品养殖户为了追求更大的商业利益和减少高密度饲养带来的风险,常常滥用抗生素药物。这种滥用抗生素的行为会引起水产品中药物的残留,而这些受污染的水产品经食物链进入人体后,容易导致蓄积毒性、细菌耐药性等一系列连锁危害,严重威胁着人们的身体健康[1]。水产品中药物的使用与残留受到越来越广泛的关注,许多国家和国际组织均制定了水产品中药物残留的限量标准。日本2006年5月29日实施的《食品中残留农药的肯定列表制度》对水产品中的134种化学药物残留量进行了限定。美国食品及药物管理局(FDA)规定对进口的水产品必须检查可能存在的221种化学药物残留和10种严禁使用的药品[2]。欧盟、加拿大、韩国等近几年也相应地提高了水产品中物残留的限量[2]。
水产品中药物残留的检测方法主要有酶联免疫法(ELISA)[3]、高效液相色谱法(HPLC)[4]、气相色谱-串联质谱法(GC-MS/MS)[5]、液相色谱-串联质谱法(LC-MS/MS)[6-8]和液相色谱-高分辨质谱法(LC-HRMS)[9]。其中,ELISA易出现假阳性结果和交叉反应;HPLC的抗干扰能力差,灵敏度低且不能满足标准限值的要求;GC-MS/MS需对样品进行繁琐的衍生化处理;LC-MS/MS无需衍生化处理,且在满足多组分同时检测的情况下仍有较好的选择性、灵敏度和特异性,弥补了前几种方法的缺陷,但其难以完全阐明化合物的结构裂解信息,定性准确度方面尚有欠缺。水产品中药物残留的前处理方法包括固相萃取、基质固相分散萃取、快速溶剂萃取、微波辅助萃取、分子印迹技术和QuEChERS等[10-15]。QuEChERS由于具有快速、简单、便宜、有效、耐用和安全可靠等优势,在检测水产品中药物残留的应用日趋增多,但是其易产生基质效应,会对方法的检出限、选择性以及测试结果的准确性产生影响。
四极杆/静电场轨道阱高分辨质谱仪(Q Orbitrap HRMS)具有分辨率高及定量能力好的优点,不同于三重四极杆低分辨质谱使用多反应监测(MRM)模式对目标物的离子对进行定量分析,Q Orbitrap HRMS可以利用目标物母离子精确的相对分子质量直接定量,无需对目标物逐个优化子离子及相关参数,对于多目标物的分析,可以极大地缩短检测时间,同时又能很好地避免低分辨质谱易受基质干扰而产生假阳性的现象[16-19]。
本研究利用改进的QuEChERS方法对水产品进行前处理,选择一种新型的高效基质脂肪吸附剂(EMR-Lipid)对样品中的杂质进行吸附,同时结合UPLC-Q Orbitrap HRMS对水产品中25种药物残留进行测定。该法灵敏、快速、简单、准确、省时、稳定,实用性强,能满足目前的检测需求。
1 实验部分
1.1 仪器、试剂与材料
Q Exactive四极杆/静电场轨道阱高分辨质谱仪(配加热电喷雾离子(HESI)源)、Dione UltiMate 3000高压液相色谱系统(美国Thermo Fisher公司); Coulter Avanti J-26XP超高速冷冻离心机(德国Bechman公司); Syncore平行定量浓缩仪、B-740再循环冷却系统、V-700/701真空泵(瑞士Buchi公司); Vortex 3涡旋振荡器(德国IKA公司); DTY-B1200电子天平(福州华志科学仪器有限公司)。
标准品:苯咪唑青霉素(纯度≥95.0%)、氯唑西林钠(纯度≥99.0%)、双钠青霉素钠盐水合物(纯度≥99.0%)、头孢噻呋(纯度≥99.0%)、苯唑西林钠(纯度≥99.0%)、哌拉西林(纯度≥98.0%)、2-氨基-5-苯并咪唑(纯度≥99.9%)、酮洛芬(纯度≥99.0%)、美洛昔康(纯度≥99.5%)、吡罗昔康(纯度≥97.0%)、水杨酸(纯度≥99.5%)、舒林酸(纯度≥98.0%)、托灭酸(纯度≥99.0%)、托美丁钠(纯度≥99.9%)、4-氨基安替比林(纯度≥99.3%)、4-甲酰氨基安替比林(纯度≥99.3%)、4-甲氨基安替比林(纯度≥98.0%)、保泰松(纯度≥99.5%)、17α-甲基睾酮(纯度≥97.6%)、醋酸美伦孕酮(纯度≥99.5%)、乙酸甲地孕酮(纯度≥99.0%)、甲羟孕酮(纯度≥99.0%)、群地龙(纯度≥98.0%)、勃地酮(纯度≥93.6%)和诺龙(纯度≥98.1%)均购自德国Dr. Ehrenstofer公司;甲醇、乙腈和无水硫酸镁(色谱纯,美国Thermo Fisher公司);高效基质脂肪吸附剂(美国Agilent公司);无水硫酸钠(Na2SO4)、氯化钠(NaCl)(分析纯,广州化学试剂厂)。实验用超纯水为Milli-Q水处理系统制得。
罗非鱼、罗氏虾、北极贝和白贝等样品由中山检验检疫局动植物监管科提供;大闸蟹购自中山华润万家超市。
1.2 标准溶液的配制
分别准确称取苯咪唑青霉素、氯唑西林、双钠青霉素、头孢噻呋、苯唑西林、哌拉西林标准品约5.0 mg,置于5 mL容量瓶中,用乙腈-水(30∶70, v/v)溶液溶解并定容至刻度,配制成质量浓度为1 g/L的标准储备液,于4 ℃冰箱中保存;分别准确称取其余标准品约5.0 mg,置于5 mL容量瓶中,用乙腈溶解并定容至刻度,配制成质量浓度为1 g/L的标准储备液,于4 ℃冰箱中保存。
准确吸取适量的上述标准储备液,置于100 mL容量瓶中,并用乙腈定容至刻度,配制成各质量浓度均为10 mg/L的混合标准工作液。
1.3 样品前处理
准确称取匀浆后的试样2.0 g(精确至0.01 g),置于25 mL具塞离心管中,加入10 mL乙腈,涡旋振荡2 min,超声提取10 min,以4 000 r/min离心5 min,移取上清液,置于用5 mL水活化过的装有EMR-Lipid 1.00 g的离心管中,涡旋振荡2 min,于5 ℃以15 000 r/min离心10 min,转移上清液至已提前放入3 g Na2SO4、3 g NaCl的25 mL离心管中,涡旋振荡2 min,于5 ℃以15 000 r/min离心10 min,将上层清液转移至15 mL离心管中,于40 ℃平行定量浓缩仪中浓缩至干,加入1.0 mL乙腈-水(10∶90, v/v)溶液,涡旋溶解残留物,提取液过0.2 μm水相滤膜,供UPLC-Q Orbitrap HRMS测定。
1.4 分析条件
1.4.1 色谱条件
色谱柱:ACQUITY UPLC BEH C18柱(100 mm×2.1 mm, 1.7 μm);柱温:40 ℃;进样量:10 μL;流动相:A相为0.1%(v/v)甲酸水溶液,B相为含0.1%(v/v)甲酸的乙腈溶液。梯度洗脱程序:0~1.0 min, 5%B; 1.0~7.0 min, 5%B~95%B; 7.0~10.0 min, 95%B; 10.0~13.0 min, 5%B。
1.4.2 质谱条件
离子源为加热电喷雾离子源;离子源温度为350 ℃;离子传输温度为320 ℃;鞘气(N2)压力为40 arb;辅助气(N2)压力为40 arb;毛细管电压为3.2 kV;离子传输管温度为325 ℃;扫描模式为全扫描/实时二级质谱(Full MS/dd-MS2)扫描;采集范围为80~1 000 Da,正负切换采集;一级质谱分辨率为70 000,二级质谱分辨率为17 500;碰撞池能量(NCE)为20、40和60 eV。25种药物的其他质谱参数见表1。
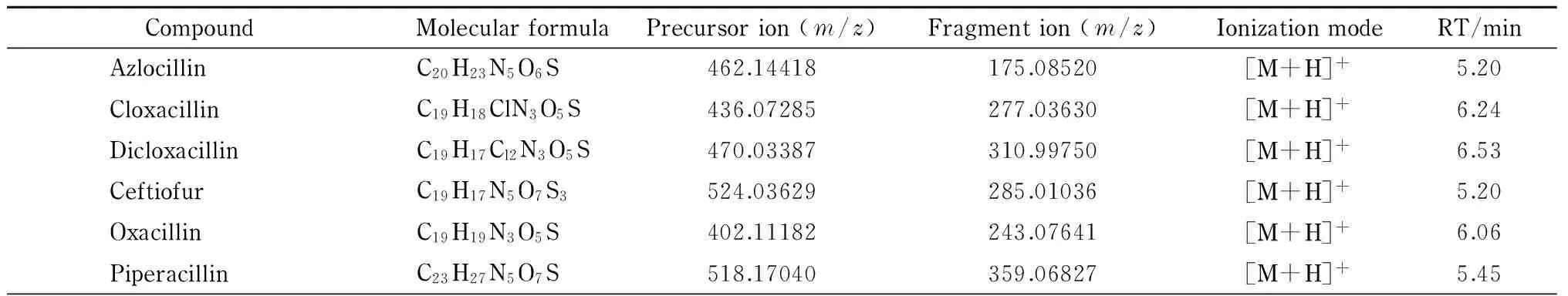
表 1 25种药物的分子式、母离子、碎片离子和保留时间Table 1 Molecular formulas, precursor ions, fragment ions and retention times (RTs) of the 25 drugs
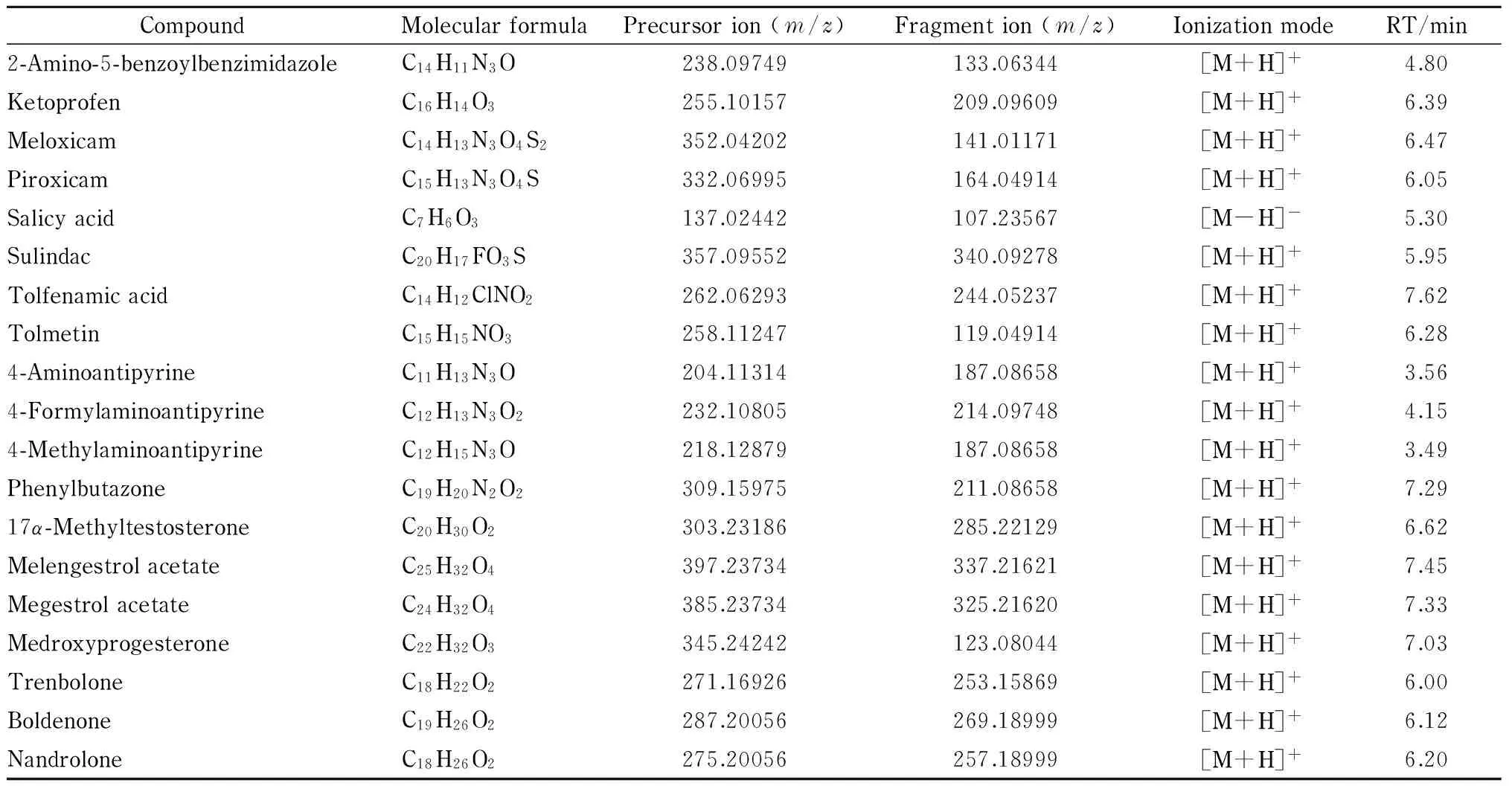
表 1 (续)Table 1 (Continued)
2 结果与分析
2.1 色谱条件的优化
本研究选择对多种农药残留分离较好的两款色谱柱Thermo Accucore RP-MS C18 (100 mm×2.1 mm, 2.6 μm)和ACQUITY UPLC BEH C18 (100 mm×2.1 mm, 1.7 μm)对25种药物进行分离。结果显示,这两款色谱柱均能分离25种药物,且峰形较好,但目标化合物乙酸甲地孕酮在ACQUITY UPLC BEH C18色谱柱上的保留时间更加稳定,质谱响应值更高,形成的色谱峰峰形尖锐,对称性好。原因可能是色谱柱粒径对乙酸甲地孕酮的分离有较大影响,粒径越小分离效果越好、越稳定。因此实验最终选用ACQUITY UPLC BEH C18色谱柱(100 mm×2.1 mm, 1.7 μm)。
本研究的25种目标物中仅有水杨酸在负离子模式下检测,其他目标物均在正离子模式下检测,为了提高正离子模式下目标物的响应值,本研究在流动相A和流动相B中均添加了甲酸,以提高目标化合物的灵敏度。比较了有机相为乙腈(含0.1%(v/v)甲酸)、水相分别为5 mmol/L乙酸铵缓冲溶液(含0.1%(v/v)甲酸)和0.1%(v/v)甲酸水溶液对25种药物色谱分离和质谱响应的影响。结果发现,使用乙腈(含0.1%(v/v)甲酸)-0.1%(v/v)甲酸水溶液为流动相时,24种药物在正离子模式下的质谱响应更高,色谱峰峰形尖锐,对称性良好;在负离子扫描模式下水杨酸的灵敏度虽有所降低,但也能够达到检测要求。因此本研究选择乙腈(含0.1%(v/v)甲酸)-0.1%(v/v)甲酸水溶液为流动相。在优化后的色谱条件下,一次进样即可完成25种目标化合物的同时分离和测定。25种药物混合标准溶液(1.0 μg/L)的选择离子色谱图见图1。

图 1 25种药物混合标准溶液(1.0 μg/L)的选择离子色谱图Fig. 1 Selected ion chromatograms of the 25 drugs in a mixed standard solution (1.0 μg/L)
2.2 质谱条件的优化
本方法以流动注射方式对25种目标物在正负切换离子模式下进行一级全扫描,采用Full MS/dd-MS2扫描模式,设定相对分子质量范围(m/z150~1 000),并以每个化合物的理论相对分子质量(见表1)建立二级扫描的目标列表。在实际扫描过程中,当一级全扫描发现目标列表里的母离子,且信号强度超过预设值,就会触发数据依赖子离子扫描模式,进而获得对应母离子精确相对分子质量的二级离子全扫描质谱信息,以实现定性确证。根据欧盟2002/657/EC对禁用药物残留检测确证方法的要求,确证检测需要4个鉴别点。本实验通过上述质谱参数的优化和筛选,每种待测物最终确定1个监测离子对(见表1),以满足欧盟兽药残留检测确证的要求。
2.3 QuEChERS条件的优化
2.3.1 萃取溶剂的优化
研究选择阴性罗非鱼肉样品为实验材料,添加含量为5.0 μg/kg的混合标准溶液,每个样品平行测定3次,外标法定量,对萃取溶剂(乙腈、含1%(v/v)甲酸的乙腈溶液和甲醇)进行优化(见图2)。结果显示,采用乙腈时,25种药物的回收率均高于70%;采用含1%(v/v)甲酸的乙腈溶液时,目标物美洛昔康等非甾体类抗炎药的回收率低于40%;采用甲醇时目标化合物的回收率普遍较低,尤其是苯咪唑青霉素等β-内酰胺类抗生素药物残留的回收率低于30%。因此本研究最终选择乙腈作为萃取溶剂。
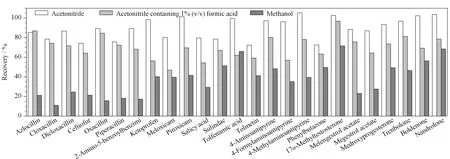
图 2 不同萃取溶剂对25种药物回收率的影响Fig. 2 Effect of different extraction solvents on the recoveries of the 25 drug

图 3 EMR-Lipid的用量对25种药物回收率的影响Fig. 3 Effect of different dosages of enhanced matrix removal of lipids (EMR-Lipid) on the recoveries of the 25 drugs
2.3.2 高效基质脂肪吸附剂用量的优化
高效基质脂肪吸附剂是一种能够选择性去除复杂基质中脂肪的独特吸附剂,具有快速、易操作的特点,对于质谱分析还具有高效的净化能力。本研究选择阴性罗非鱼样品为实验材料,添加含量为5.0 μg/kg的混合标准溶液,每个样品平行测定3次,外标法定量,对EMR-Lipid的使用量进行优化(见图3)。在优化范围(0.2~1.5 g)内,当添加量为1.0 g时25种药物残留的回收率均能达到78%以上;继续增加EMR-Lipid的使用量,虽然有的药物加标回收率有所提高,但并不显著,有的药物加标回收率反而下降,主要原因是EMR-Lipid使用量增加到一定量时会对部分药物产生吸附作用。研究最终确定EMR-Lipid的使用量为1.0 g。
2.3.3 离心条件和浓缩条件的优化
虽然采用EMR-Lipid已经去除了样品中大部分的油脂,但是样品提取液中还残留少量油脂、蛋白质等干扰组分。本研究采用高速冷冻离心对样品提取液进行处理,能够有效地促进油脂凝结和蛋白质的沉降,减少其对目标化合物的干扰。
本研究对样品提取液进行了浓缩,选择平行定量浓缩仪减压浓缩样品提取液,并对浓缩的条件进行摸索,确定浓缩温度为40 ℃,冷凝温度为-2 ℃,真空度采用梯度下降的方式(250 kPa 10 min, 80 kPa 10 min, 30 kPa浓缩至干)。该方法可以同时浓缩处理24份样品提取液,完全浓缩至干仅需1.5 h。该处理方法能够有效降低检测成本,提高检测质量和效率。
2.3.4 净化萃取剂和使用量的选择
本研究考察了无水硫酸钠与氯化钠、无水硫酸镁与氯化钠作为净化萃取剂。结果显示,当选择无水硫酸镁与氯化钠作为净化萃取剂时,由于无水硫酸镁对目标化合物有较强的吸附作用,25种药物的回收率均小于20%;当选择无水硫酸钠与氯化钠作为净化萃取剂时,25种药物的回收率均高于70%。研究选择阴性罗非鱼样品为实验材料,添加含量为5.0 μg/kg的混合标准溶液,每个样品平行测定3次,外标法定量,对无水硫酸钠和氯化钠的使用量进行优化(见图4)。无水硫酸钠和氯化钠的组合分别为1 g Na2SO4+5 g NaCl、2 g Na2SO4+4 g NaCl、3 g Na2SO4+3 g NaCl、4 g Na2SO4+2 g NaCl和5 g Na2SO4+1 g NaCl,研究结果显示,当Na2SO4和NaCl均为3 g时,目标物的回收率最高。因此本研究选择3 g Na2SO4和3 g NaCl作为净化萃取剂。
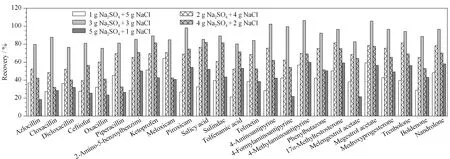
图 4 不同净化萃取剂对25种药物回收率的影响Fig. 4 Effect of different cleaning extraction agents on the recoveries of the 25 drugs
2.4 方法学验证
2.4.1 标准曲线、线性范围和检出限
在高分辨质谱的分析中,其提取的精确相对分子质量色谱图无基线噪音,因此采用传统信噪比(S/N)的方法无法计算检出限。本方法采用基质加标的方式建立标准曲线,以定量离子的峰面积(Y)为纵坐标、相应质量浓度(X, μg/L)为横坐标绘制标准曲线。用标准曲线各浓度点偏离值(实际值与理论值之差)的标准偏差的3.3倍以上的质量浓度确定检出限[9]。准确量取适量混合标准工作液,用乙腈-水(10∶90, v/v)溶液稀释成系列梯度的标准工作液,在1.4节条件下依次测定。结果显示,目标化合物在各自的范围内线性关系良好,相关系数(r)均大于0.997,检出限为0.1~1.0 μg/kg(见表2)。

表 2 25种药物的线性范围、回归方程、相关系数和检出限Table 2 Linear ranges, regression equations, correlation coefficients (r) and LODs of the 25 drugs

表 2 (续)Table (Continued)
Y: peak area;X: mass concentration, μg/L.
2.4.2 准确度和精密度
在阴性水产品样品(包括罗非鱼、罗氏虾、北极贝和大闸蟹)中进行加标回收试验。分别对上述样品进行3个水平的加标回收率测定,每个水平进行6次平行试验,结果表明,25种药物的平均加标回收率为70.1%~108.9%,RSD为2.1%~13.8%(见表3)。

表 3 实际空白样品中25种药物的加标回收率和相对标准偏差(n=6)Table 3 Spiked recoveries and RSDs of the 25 drugs in real blank samples (n=6)
表 3 (续)Table 3 (Continued)
2.4.3 实际样品检测
利用本研究建立的分析方法检测37批实际样品(其中包括24批罗非鱼样品、7批罗氏虾样品和6批白贝样品),其中在2批罗非鱼样品中分别检出苯咪唑青霉素和美洛昔康,检出含量分别为6.92 μg/kg和2.64 μg/kg,其他样品中均无药物残留检出。实际样品中苯咪唑青霉素和美洛昔康的二级质谱图分别见图5和图6。

图 5 苯咪唑青霉素(a)阳性样品(6.92 μg/kg)和(b)标准溶液(5.0 μg/L)的二级质谱图Fig. 5 MS2 spectra of azlocillin in (a) a positive sample (6.92 μg/kg) and (b) a standard solution (5.0 μg/L)
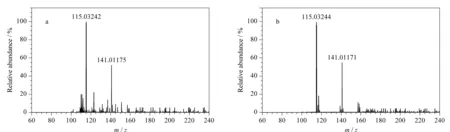
图 6 美洛昔康(a)阳性样品(2.64 μg/kg)和(b)标准溶液(1.0 μg/L)的二级质谱图Fig. 6 MS2 spectra of meloxicam in (a) a positive sample (2.64 μg/kg) and (b) a standard solution (1.0 μg/L)
3 结论
本研究建立了UPLC-Q Exactive Orbitrap HRMS检测水产品中25种药物残留的分析方法,以乙腈为提取剂,水产品样品经改进的QuEChERS方法进行净化处理,以EMR-Lipid去除提取液中的干扰物质,无水硫酸钠与氯化钠为盐析剂反相萃取目标化合物。该法满足GB/T 27404-2008《实验室质量控制规范食品理化检测》的技术要求。
[1] Pan M F, Wang J P, Fang G Z, et al. Food Science, 2014, 35(15): 277
潘明飞, 王俊平, 方国臻, 等. 食品科学, 2014, 35(15): 277
[2] Sun W H, Xing L H, Zhai Y X, et al. Journal of Food Safety and Quality, 2014, 5(1): 14
孙伟红, 邢丽红, 翟毓秀, 等. 食品安全质量检测学报, 2014, 5(1): 14
[3] Guo H, Wang Y F, Zou M Q, et al. Food Science, 2012, 33(20): 144
郭浩, 王燕飞, 邹明强, 等. 食品科学, 2012, 33(20): 144
[4] Liang X H, Dai Z Y, Liu W E. Journal of Central South University: Medical Science, 2009, 34(7): 689
梁湘辉, 戴智勇, 刘文恩. 中南大学学报: 医学版, 2009, 34(7): 689
[5] Wang L P, Li X, Sun Y, et al. Chinese Journal of Analytical Chemistry, 2005, 33(7): 951
汪丽萍, 李翔, 孙英, 等. 分析化学, 2005, 33(7): 951
[6] Li N, Zhang Y T, Liu L, et al. Chinese Journal of Chromatography, 2014, 32(12): 1313
李娜, 张玉婷, 刘磊, 等. 色谱, 2014, 32(12): 1313
[7] Yang W H, Wu S M, Cai X M, et al. Chinese Journal of Chromatography, 2017, 35(10): 1062
杨旺火, 吴少明, 蔡小明, 等. 色谱, 2017, 35(10): 1062
[8] Zhang X C, Hua H B, Zhang P J, et al. Food Science, 2012, 33(12): 190
张宪臣, 华洪波, 张朋杰, 等. 食品科学, 2012, 33(12): 190[9] Guo M M, Guo J, Wu H Y, et al. Chinese Journal of Analytical Chemistry, 2016, 44(10): 1504
郭萌萌, 国佼, 吴海燕, 等. 分析化学, 2016, 44(10): 1504
[10] Sun X Q, Dong Z L, Li Y C, et al. Transactions of the Chinese Society of Agricultural Engineering, 2014, 30(8): 280
孙兴权, 董振霖, 李一尘, 等. 农业工程学报, 2014, 30(8): 280
[11] Zou Y, Shao L Z, Chen S M, et al. Chinese Journal of Chromatography, 2017, 35(8): 801
邹游, 邵琳智, 陈思敏, 等. 色谱, 2017, 35(8): 801
[12] Zhang K M, Liang F Y, Deng M, et al. Chinese Journal of Chromatography, 2016, 34(9): 860
张科明, 梁飞燕, 邓鸣, 等. 色谱, 2016, 34(9): 860
[13] Gonzalez-Curbelo M A, Socas-Rodriguez B, Herrera-Herrera A V, et al. TrAC-Trends Anal Chem, 2015, 1: 169
[14] Rejczak T. AOAC Int, 2015, 98: 1143
[15] Han L, Matarrita J, Sapozhnikova Y, et al. J Chromatogr A, 2016, 1449: 17
[16] Jia W, Chu X G, Ling Y, et al. J Chromatogr A, 2014, 1347: 122
[17] Solliec M, Roy-Lachapelle A, Sauve S. Anal Chim Acta, 2015, 853: 415
[18] Park J A, Zhang D, Kim D S, et al. J Sep Sci, 2015, 38(16): 2772
[19] Geis-Asteggiante L, Lightfield A R, Dutko T, et al. J Chromatogr A, 2012, 1258: 43
