基于Egfp启动子高通量筛选方法的研究
2018-03-01徐佳皮莉张玉崔丹瑶张翠英肖冬光
徐佳,皮莉,张玉,崔丹瑶,张翠英,肖冬光
(天津市工业微生物重点实验室,天津科技大学生物工程学院,天津 300457)
绿色荧光蛋白(Gfp)是一类存在于水母、水螅和珊瑚等腔肠动物体内的生物发光蛋白[1]。当用395 nm的紫外线或475 nm的蓝光激发,Gfp就可在508 nm处发射绿色荧光。由于其基因可以在异源组织中表达并产生荧光[2],检测时不需抗体、辅因子、酶底物等其它成分,不影响宿主细胞[3],因而可以鉴定、跟踪、分选表达Gfp的活细胞,Gfp近年来成为细胞生物学和分子生物学中广泛应用的报告蛋白[4]。Gfp已广泛应用于报告蛋白、融合标记、生物传感器、pH检测传感器、蛋白间相互作用、胞内各器官蛋白的传递等领域。而与其它常用的报告基因相比,Gfp具有性质稳定、无细胞毒性、灵敏性、检测便捷等优点[5],在荧光显微镜下,可直接观察到活细胞中的Gfp绿色荧光,因此可作为研究与之融合的其他蛋白表达的报告基因。
GFPS65T突变体(S65突变为T)荧光强度比野生型Gfp强5倍,进一步进行F64L突变得到37 ℃下高效成熟的增强型绿色荧光蛋白(Egfp),大大增强了该报告分子的灵敏度[6],是目前应用最广泛的荧光蛋白之一。但Egfp一般都需强启动子以驱动Gfp基因在细胞内足量的表达[7],多数生物具有微弱的自发荧光现象,并有类似的激发和发射波长,影响了某些Gfp的检测[8]。
近年来,由一系列具有不同调控强度的突变启动子构成的启动子文库,被用于对目的基因的表达进行精确调控。启动子文库是不同启动子强度的集合体,在构建时,可对天然启动子序列进行突变,再从中筛选出合适强度的多个突变启动子。
构建过程中,启动子序列的突变方式多种多样,如易错PCR[9],DNA shuffling[10],定点突变和化学合成[11]等,已基本成为常规操作,只要对反应条件稍作优化,即可获得包含足够丰富的突变序列。
Alper[12]等人通过易错PCR,引入Gfp基因,获得了噬菌体启动子活性196倍范围内的突变启动子文库,证明可通过易错PCR获得启动子活性差别较大的突变体,实现了对基因的精细调控。秦秀林[14]等人建立了适用于毕赤酵母组成型表达文库筛选的高通量方法,利用高通量方法筛选了300株整合了(PGAP突变序列-yEGFP)表达单元的重组菌,从中挑选出6株yEGFP表达水平在G/GHg 0.6~218%范围内的重组细胞(G01、G02、G03、G0、G6)。因此启动子文库构建的难点不在于如何实现启动子突变序列的多样性,而在于建立一个高效、灵敏和可靠的高通量筛选方法[13],能有效地筛选到强度改变的启动子突变体。故本研究以筛选PGK突变子为目标,通过优化培养条件,建立可靠的高通量筛选系统,为构建PGK启动子文库奠定基础。
1 材料与方法
1.1 材料与试剂
1.1.1 菌株
工业酿酒酵母α5(酿酒酵母工业菌株AY15的α型单倍体)、大肠杆菌(Escherichia coli)DH5α、PyEGFP3质粒、PAXSK质粒、pUC-PIAK质粒,以上均由天津科技大学天津市工业微生物重点实验室保藏。
1.1.2 培养基和溶液
YEPD培养基:葡萄糖2%,蛋白胨2%,酵母浸粉1%,pH自然,115 ℃高压蒸汽灭菌20 min。固体培养基需再添加2%琼脂粉。
LB培养基:胰蛋白胨1%,酵母浸粉0.5%,NaCl 1%,pH自然,115 ℃高压蒸汽灭菌20 min。固体培养基需再添加2%琼脂粉。
MD培养基:YNB 13.4 g/L,生物素0.4 mg/L,葡萄糖20 g/L,pH 6.0,115 ℃,0.1 MPa灭菌30 min。
SD培养基:YNB 0.67%,葡萄糖2%,pH 6.0,115 ℃,0.1 MPa灭菌30 min。
BMD培养基:1M K2HPO4(pH 6.0)100 mL/L,YNB 13.4 g/L,生物素0.4 mg/L,葡萄糖20 g/L,pH 6.0,115 ℃、0.1 MPa灭菌30 min。
BMDY培养基:1M K2HPO4(pH 6.0)100 mL/L,YNB 13.4 g/L,生物素0.4 mg/L,葡萄糖20 g/L,酵母浸粉10 g/L。pH 6.0,115 ℃、0.1 MPa灭菌30 min。
BSM1培养基:(NH4)2SO47 g/L,CaSO40.46 g/L,K2SO49.1 g/L,MgSO4·7H2O 7.5 g/L,PTM1 12 mL/L,葡萄糖20 g/L。pH 6.0,115 ℃、0.1 MPa灭菌30 min。
微量元素PTM1:CuSO4·5H2O 6 g/L,KI 0.08 g/L,生物素0.2 g/L,MnSO4·H2O 3 g/L,FeSO4·7H2O 65 g/L,ZnSO4·7H2O 20 g/L,CoCl2·6H2O 0.5 g/L,H3BO30.02 g/L。
PBS缓冲溶液:KH2PO40.27 g/L,Na2HPO41.42 g/L,NaCl 8 g/L,KCl 0.2 g/L。用去离子水定容至1 L,用HCl/NaOH调pH 7.4,4 ℃冰箱保存。
1 mol/L醋酸锂溶液:准确称取6.6 g醋酸锂溶于80 mL蒸馏水中,并定容至100 mL,过膜除菌或121 ℃高压蒸汽灭菌15 min,4 ℃冰箱保存。
1.1.3 酶与化学试剂
酵母基因组提取试剂盒和质粒提取试剂盒购自Solarbio公司;限制性内切酶、T4 DNA连接酶、Pfu DNA聚合酶和DNTP购自宝生物工程(大连)有限公司;PCR产物回收试剂盒(Cycle Pure Kit)购自Omega公司。
1.2 仪器与设备
M200PRO型光栅型多功能酶标仪:瑞士帝肯有限公司;BX53F型荧光显微镜:日本OLYMPUS会社;岛津紫外分光光度计UVmini-1240:岛津仪器(苏州)有限公司;F0199型48孔细胞培养板:美国Coring公司;F0199型96孔酶标板:美国Coring公司。
1.3 方法
1.3.1 引物设计与合成
本实验所用引物由北京鼎国昌盛生物技术有限责任公司合成,酶切位点以下划线标注。

表1 本实验所用引物Table 1 Primers used in the current study
1.3.2 pUC-PEBBK质粒的构建
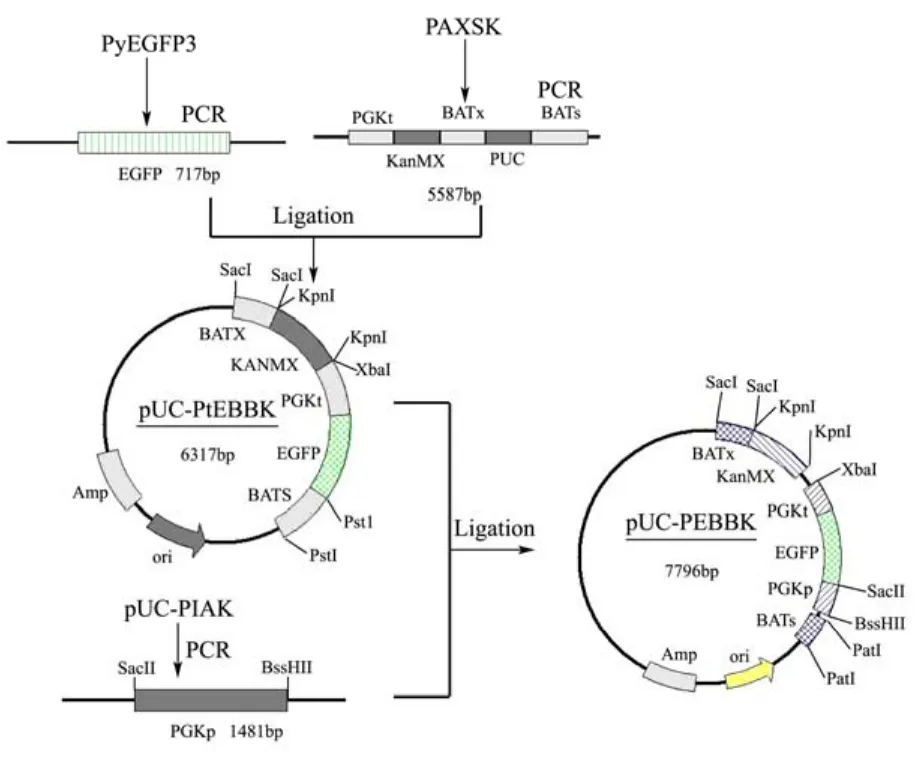
图1 重组质粒pUC-PEBBK构建流程图Fig.1 Construction flowchart of recombinant plasmid pUC-PEBBK
质粒构建流程图如图1所示。以PyEgfp3质粒为模板,分别以EGFP-U2/EGFP-D1和EGFP-U1/EGFP-D2为引物进行PCR扩增,得到EGFP基因片段,反应条件:95 ℃ 5 min;94 ℃ 1 min;56 ℃ 1 min;72 ℃ 2 min;循环30次,72 ℃ 10 min。PCR产物按照PCR产物回收试剂盒说明书的要求进行纯化回收。以实验室保存的PAXSK质粒为模板,分别以PA-U1/PA-D2和PA-U1/PA-D2为引物进行PCR扩增,得到PGKt-KanMX-BATx-pUC-BATs基因片段,纯化回收连接,得到pUC-PtEBBK质粒。以实验室保存的pUC-PIAK质粒为模板,以PGKp-U和PGKp-D为引物进行PCR扩增,得到PGKp基因片段,将PtEBBK质粒用SacII和BssHII酶切线性并去磷酸化,与同样经过SacII和BssHII酶切的PGKp基因片段连接,得到pUC-PEBBK质粒。
1.3.3 突变菌株α5-PEBBK的构建
酵母以质粒pUC-PEBBK为模板,BATs-U和BATx-D为引物PCR扩增基因片段。将纯化后的重组片段BATs-PGKp-Egfp-PGKt-KanMX-BATx,经醋酸锂化学转化法[15]将其导入到酿酒酵母单倍体α5中,并使重组菌株产生G418抗性,使用含G418 YEPD平板进行筛选。
1.3.4 荧光显微镜检测
将重组菌株α5-PEBBK用YEPD培养基培养12 h,将菌液离心,收集菌体,用PBS(pH 7.0)稀释至OD600=1,取一滴制成玻片,使用荧光显微镜检测细胞Egfp荧光强度,通过荧光显微镜观察。
1.3.5 酶标仪检测
重组菌株α5-PEBBK 和亲本菌株α5经YPD培养后,菌体收集用PBS洗一次,用PBS(pH 7.0)稀释至OD600=1,经PBS重悬后将稀释后样品取250 μL至96孔板,使用荧光酶标仪检测Egfp荧光强度(激发波长488 nm,发射波长510 nm),并检测OD600,检测Egfp荧光强度时,以不表达Egfp的基因工程菌α5作为对照去除背景干扰,比荧光强度(RFU/OD600)为荧光强度值比上对应细胞密度。
1.3.6 培养基对重组菌生长的影响
分别用SD、MD、BMD、BSM1培养基培养重组菌α5-PEBBK和亲本菌株α5,各作三个平行,经30 ℃,200 r/min,试管培养36 h,测定细胞密度OD600。
1.3.7 培养基对Egfp表达的影响分别用SD、MD、BMD、BSM1培养基培养重组菌α5-PEBBK和亲本菌株α5,经30 ℃,200 r/min,试管培养36 h,经荧光酶标仪检测荧光强度RFU,根据1.3.6测得的细胞密度OD600,计算比荧光强度
RFU/OD600。
1.3.8 48孔板接种量的优化
在确定了合适的培养基后,利用48孔深孔板,对不同阶段的接种量和培养时间进行优化,每孔装900 μL BSM1,分别取10 μL、20 μL、30 μL、40 μL经过24 h培养的种子液,30 ℃,200 r/min培养60 h,每间隔12 h取样测细胞密度OD600。
1.3.9 最佳检测荧光强度时间的确定
用BSM1培养基培养重组菌α5-PEBBK和亲本菌株α5,各接种40 μL种子液,30 ℃,200 r/min培养60 h,每间隔12 h取样测OD600和RFU,计算比荧光强度RFU/OD600。
2 结果与讨论
2.1 增强型绿色荧光蛋白Egfp在酿酒酵母中的表达
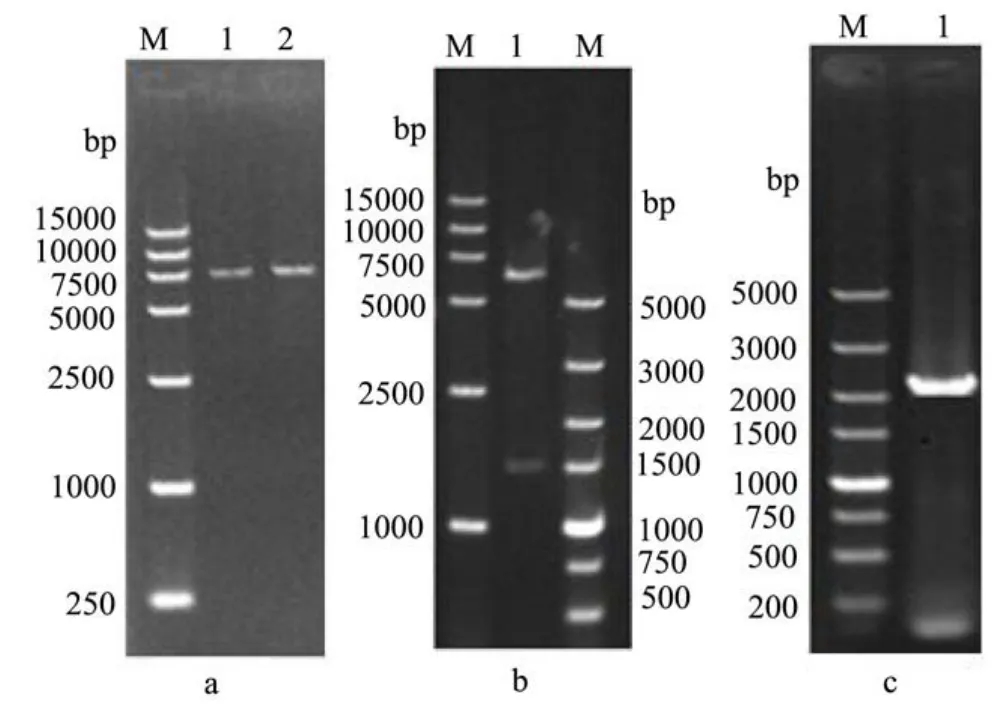
图2 重组质粒pUC-PEBBK验证Fig.2 The confirmation of the recombinant plasmid pUC-PEBBK
2.1.1 pUC-PEBBK质粒的验证和BssHII双酶切后。(c)M:DL5000 DNA Marker;1:BATs-U和EGFP-D为引物PCR验证。
按照1.3.2方法获得重组质粒,提取质粒pUC-PEBBK,将重组质粒pUC-PEBBK分别用SacII和BssHII单酶切,酶切后得到7785 bp的单一条带,如图2a。分步用SacII和BssHII双酶切后得到6300 bp和1481 bp的条带(图2b),证明连接正确。并用引物PGKp-U和EGFP-D PCR验证,得到2300 bp大小正确条带,结果如图2c。
2.1.2 重组菌株α5-PEBBK的验证

图3 α5-PEBBK上下游定点验证Fig.3 The PCR verification of the mutant strain α5-PEBBK
将转化子提取基因组DNA,然后PCR定点验证,结果如图3所示。以重组菌株基因组DNA为模板,亲本菌株α5基因组DNA为阴性对照,分别以B1-U和PGKp-D,PGKp-U和B2-D两对引物进行上下游定点验证,阴性对照α5亲本菌株由于不含有Egfp基因及其与PGK启动子相连序列,无法扩增出目的条带,重组菌株则可以扩增出2456 bp和4028 bp大小条带。由图可知,PCR目的条带单一,大小正确,证明BAT2基因已经敲除,基因整合成功,得到正确的重组菌株α5-PEBBK。
2.1.3 荧光显微镜检测Egfp荧光

图4 荧光显微镜检测重组菌株α5-PEBBK Egfp荧光强度Fig.4 Fluorescence of Egfp measured by the fluorescence microscope in α5-PEBBK strain
使用荧光显微镜检测重组细胞Egfp荧光强度,在重组菌细胞内观察到Egfp表达所产生的荧光(图4),结果证明启动子PGK可调控绿色荧光蛋白Egfp在酵母中的表达。
2.1.4 多功能酶标仪检测Egfp荧光强度
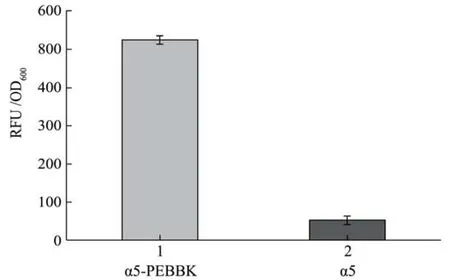
图5 酶标仪检测重组菌株α5-PEBBK与亲本菌株α5Egfp荧光强度Fig.5 Fluorescence of Egfp measured by the fluorescence microplate in α5-PEBBK and α5 strains
利用多功能酶标仪检测重组细胞Egfp荧光强度,如图5所示,α5-PEBBK和α5的比荧光强度值分别为523和53,α5-PEBBK的比荧光强度是亲本菌株α5的10倍。
2.2 PGK启动子文库高通量筛选方法的建立
2.2.1 培养基优化
2.2.1.1 培养基对Egfp荧光检测的干扰
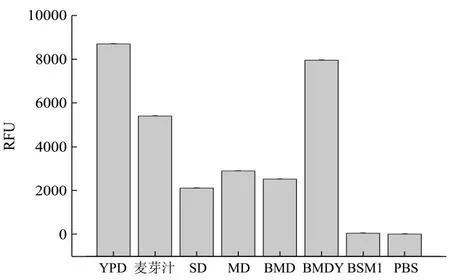
图6 荧光酶标仪测定无菌培养基荧光强度Fig.6 The fluorescence of the aseptic medium measured by fluorescence microplate
为了排除培养基成分对荧光检测的干扰,以PBS为阴性对照,考察不同培养基对Egfp荧光检测的影响,结果如图6。
BSM1培养基的本底荧光强度值最小(51 RFU),与PBS(54 RFU)接近,其次是SD(2100 RFU),MD(2910 RFU),BMD(2531 RFU),无菌培养基BMDY和YPD经荧光酶标仪检测荧光强度最强,分别为:7971 RFU和8717 RFU,与PBS相比,高出近150倍。
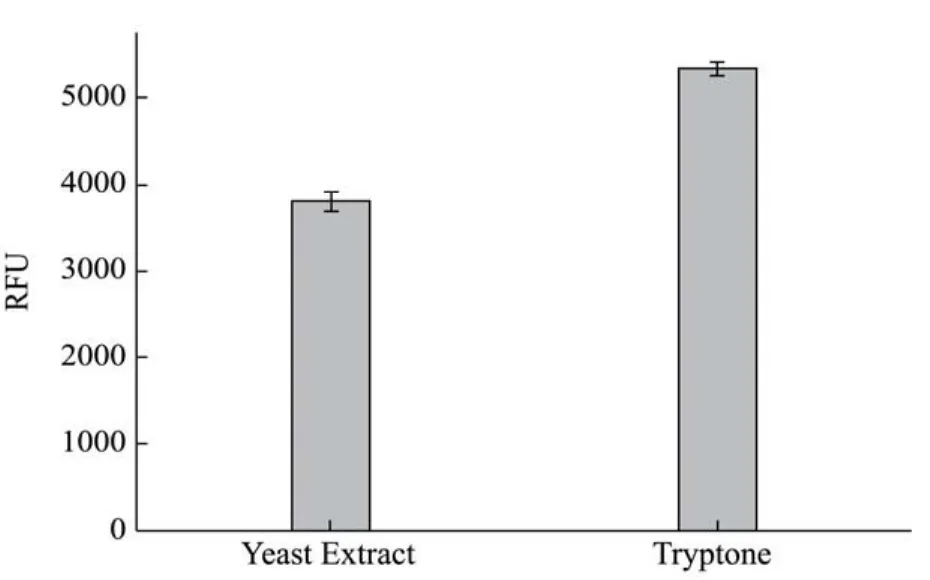
图7 荧光酶标仪测定酵母浸粉(Yeast Extract)和蛋白胨(Tryptone)荧光强度Fig.7 The fluorescence of yeast extract and tryptone measured by fluorescence microplate
在本实验中,发现培养基成分对Egfp的检测也有干扰,通过比较分析这几个培养基成份,干扰Egfp的荧光检测可能的物质:培养基BMDY和YPD中含有酵母浸粉(Yeast Extract)和蛋白胨(Tryptone),通过荧光酶标仪检测,2% Yeast Extract和1% Tryptone溶液的荧光强度分别为3800 RFU和5400 RFU(图7),证明含有酵母浸粉和蛋白胨的培养基均不适合检测荧光菌株的培养。
2.2.1.2 培养基对重组菌生长的影响
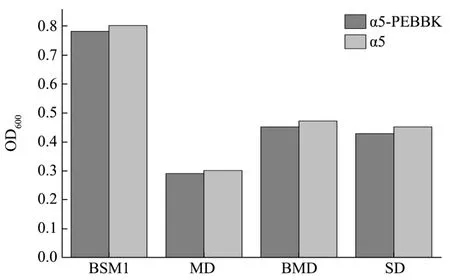
图8 不同培养基对重组菌生长的影响Fig.8 The effects of medium on the recombinant straingrowth
鉴于BSM1、SD、MD和BMD培养基对Egfp荧光强度测定干扰较小,可作为筛选用的培养基,又进一步考查了它们对重组菌生长影响(图8)。
结果表明,重组菌α5-PEBBK和亲本菌株α5在同一种培养基上的生长速度没有明显差异,相比BMD、SD、MD三种培养基,BSM1更有利于两株菌生长,两种菌的细胞密度OD600均超过它们在BMD、SD、MD中的细胞密度。
2.2.1.3 培养基对重组菌Egfp表达影响
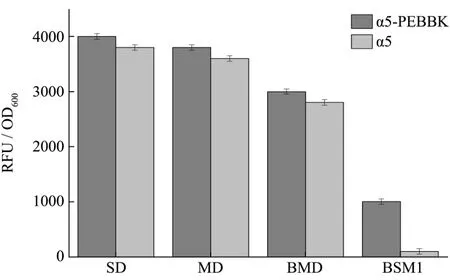
图9 不同培养基对重组菌Egfp表达影响Fig.9 The effects of medium on the Egfp expression
结果表明,重组菌α5-PEBBK经SD、MD、BMD培养基培养后,Egfp比荧光强度与亲本菌株相差不大,而在BMS1中培养,重组菌株比荧光强度为937,是亲本菌株比荧光强度的10倍(图9)。综上所述,在重组细胞α5-PEBBK中,基因Egfp在启动子PGK的调控下获得了正确的表达,但由于仅有1个拷贝基因整合在重组菌的染色体中,通过利用荧光酶标仪检测不同培养基对Egfp荧光表达的影响,发现在YPD、BMDY、麦芽汁培养基中Egfp表达产生的荧光强度受到较强干扰,在SD、MD、BMD培养基中生长缓慢,在BSM1培养基中受到干扰较弱,便于检测,比荧光强度重组菌和亲本菌株相差较大,且较利于其生长,因此选定BSM1培养基为筛选培养基。
2.2.2 筛选条件的优化
2.2.2.1 接种量的确定
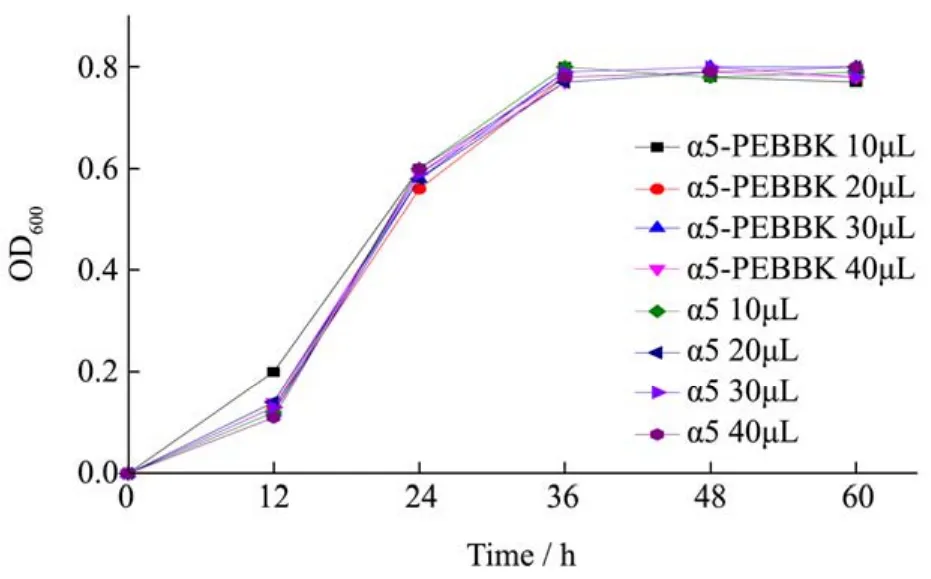
图10 培养板中不同接种量对重组菌和对照菌生长影响Fig.10 The effects of inoculum volume on the strains growth of α5-2 and α5 in the plate
由图10可以看出,即使初始接种量不同,但重组菌α5-PEBBK和原菌α5生长曲线趋势保持一致,培养板接种量对最终细胞密度无明显影响,接种量为10 μL、20 μL、30 μL、40 μL时,重组菌在培养36 h 后,都从对数生长期过渡至稳定期,培养板培养时间36 h细胞密度最大,达到平稳期。但考虑到较大的接种量可降低操作实验误差,因此,确定预培养板的接种量为40 μL。
2.2.2.2 最佳检测荧光强度时间的确定
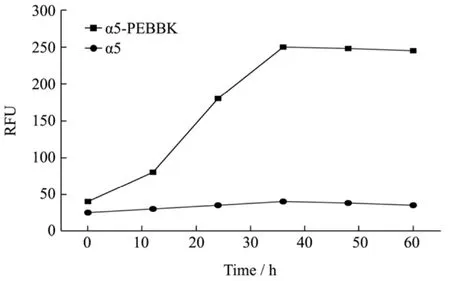
图11 重组菌荧光强度与时间的关系Fig.11 The correlation between fluorescence and time of recombinant bacteria

图12 重组菌不同培养时间的比荧光强度Fig. 12 Specific fluorescence of recombinant strains at different time points
培养36 h重组菌与亲本菌株荧光强度差异最大,如图11,对重组菌α5-PEBBK而言,随着时间的增加,荧光强度也线性增加,当培养36 h以后,荧光强度的增长变缓;而随着时间的增加,亲本菌株α5的荧光强度始终恒定在一较低水平,实验结果说明在36 h,α5-PEBBK与亲本菌株α5荧光强度差异最大。通过考察不同时间点重组菌的比荧光强度,如图12,可以发现,36~60 h重组菌比荧光强度维持在一恒定水平,因此,确定培养板培养36 h后测定重组菌Egfp荧光强度。
3 结论
3.1 在本研究中,我们以pUC19为载体,磷酸甘油酸激酶基因PGK启动子为上游调控元件,KanMX抗性基因为筛选标记,构建酵母表达质粒pUC-PEBBK,将绿色荧光蛋白Egfp整合到酿酒酵母基因组上,在启动子调控下表达,同时敲除BAT2基因,获得重组菌株α5-PEBBK,为筛选突变启动子作为亲本菌株,通过荧光显微镜可以清晰的看到Egfp基因在酵母细胞内的表达。荧光分光光度计测得其激发波长488 nm,吸收波长510 nm,荧光酶标仪检测重组菌α5-PEBBK荧光强度是亲本菌株α5的10倍。Egfp能够在启动子PGK调控下在酿酒酵母中表达。
3.2 利用48深孔板结合荧光酶标仪检测,建立了启动子文库的高通量筛选方法,通过培养基优化,排除了YPD、BMDY、麦芽汁培养基中蛋白胨和酵母浸粉对绿色荧光蛋白检测的干扰,实验结果显示在SD、MD、BMD中酿酒酵母生长缓慢,最终确定了利于Egfp表达和快速检测的筛选培养基BSM1,最佳48孔板培养条件为接种量40 μL,培养时间为36 h。研究结果为实现48孔板高效、灵敏、可靠地筛选到强度微量改变的启动子突变体奠定基础。
[1] Shimomura O, Johnson F H, Saiga Y. Extraction, purification,and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea [J]. J Cell Comp. Physiol.,1962, 59(3): 223-239
[2] 刘歆.绿色荧光蛋白基因(gfp)转化柑橘及植株再生[D].武汉:华中农业大学,2005
LIU Xin. Transformation of green fluorescent protein gene(gfp) into citrus species and transgenic plants regeneration[D]. Wuhan: Huazhong Agricultural University, 2005
[3] 吕晓杰,周广东,马俐君,等.增强型绿色荧光蛋白转染活细胞效率的研究[J].组织工程与重建外科,2011,7(5):248-253
LV Xiao-jie, ZHOU Guang-dong, MA Li-jun, et al. The effectiveness of labeling technique of enhanced green fluorescent protein by transfection into living cell [J]. Journal of Engineering and Reconstructive Surgery, October, 2011,7(5): 248-253
[4] 刘伟,林志伟,陈美霞,等.枯草芽孢杆菌绿色荧光蛋白高效表达载体的构建[J].热带作物学报,2012,33(3):467-471
LIU Wei, LIN Zhi-wei, CHEN Mei-xia, et al. Construction of high-level expression of green fluorescent protein vector in Bacillus subtilis [J]. Chinese Journal of Tropical Crops, 2012,33(3): 467-471
[5] 侯清华,宋淑亮,梁浩,等.增强型绿色荧光蛋白的色谱分离和纯化[J].色谱,2013,31(2):151-154
HOU Qing-hua, SONG Shu-liang, LIANG Hao, et al.Isolation and purification of enhanced green fluorescent protein using chromatography [J]. Chinese Journal of Chromatography, 2013, 31(2): 151-154
[6] Heim R, Cubitt A B, Tsien R Y. Improved green fluorescence[J]. Nature, 1995, 6516: 663-664
[7] Chalfie M, Tu Y, Euskirchen G, et al. Green fluorescent protein as a maker as a maker for gene expression [J].Science, 1994, 263: 802-805
[8] 杨明瑜,刘翔.利用绿色荧光蛋白GFP作为报告基因检测转基因植物[J].食品与生物技术学报,2008,31(1):96-102
YANG Ming-yu, LIU Xiang. Using green fluorescent protein as a reporter gene to detect transgenic plant [J]. Journal of Food Science and Biotechnology, 2008, 31(1): 96-102
[9] Ellis T, Wang X, Collins J J. Diversity-based, model-guided construction of synthetic gene networks with predicted functions [J]. Nat. Biotechnol., 2009, 27(5): 465-471
[10] Lu C, Jeffries T. Shufflling of promoters for multiple genes to optimize xylose fermentation in an engineered Saccharomyces Cerevisiae strain [J]. Appl. Environ.Microbiol., 2007, 73: 6072-6077
[11] Bakke I, Berg L, Aune T. E, et al. Random mutagenesis of the PM promoter as a powerful strategy for improvement of recombinant-gene expression [J]. Appl. Environ. Microbiol.,2009, 75: 2002-2011
[12] Alper H, Fischer C, Nevoigt E, et al. Tuning genetic control through promoter engineering [J]. Proc. Natl. Acad. Sci.USA., 2005, 102(36): 12678-12683
[13] Tyo K E, Alper H S, Stephanopoulos G N. Expanding the metabolic engieering toolbox: more options to engineer cells[J]. Trends Biotechnol., 2007, 25(3): 132-137
[14] 秦秀林.构建GAP启动子文库提高重组毕赤酵母S-腺苷甲硫氨酸合成[D].上海:华东理工大学,2011
QIN Xiu-lin. Construction of functional GAP promoter library pave the way for metabolic engineering of S-Adenosylmethionine production in pichia pastoris [D].Shanghai: East China University of Science and Technology,2011
[15] Gietz R D, Woods R A. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method [J]. Methods in Enzymology, 2002, 350: 87-96
