反相高效液相色谱法测定硫酸西索米星氯化钠注射液的含量
2018-02-15侯玉荣范青峰史孙亮袁耀佐
侯玉荣,范青峰,史孙亮,袁耀佐,张 玫
(1江苏省食品药品监督检验研究院,南京 210008;2中国药科大学,南京 210009)
西索米星(sisomicin)是20世纪60年代开发的由小单孢菌产生的首个含双键的氨基糖苷类抗生素[1-2],也是奈替米星的合成用母体[3],结构与庆大霉素相似,其硫酸盐制剂在临床上被广泛应用,具有疗效好、价格较低等特点。国家食品药品监督管理局网站上显示,目前我国硫酸西索米星原料有4个厂家生产,注射液、注射用粉针等不同剂型有32个生产厂家,原料药质量标准收载入中国药典、日本药局方、美国药典、韩国药典[4-7],除《中华人民共和国药典》采用HPLC-UV法测定原料的含量外,其余均采用抗生素微生物检定法。硫酸西索米星氯化钠注射液质量标准收载入《国家食品药品监督管理局标准》,采用抗生素微生物检定法测定含量。
抗生素微生物检定法是一种生物活性测定法,通过测定供试品抗生素抑菌圈的直径大小,与标准物质进行比较,计算含量,以效价单位作为计量单位,缺点是操作复杂、影响因素多、误差较大、实验时间长等;目前美国药典、日本药局方和韩国药典均采用抗生素微生物检定法控制硫酸西索米星原料药的含量,仅中国药典采用HPLC-UV法控制原料药的含量,但中国药典的HPLC-UV法主成分与前杂质分离不理想。本研究在目前已有各方法的基础上,建立了一种专属性更好的反相高效液相色谱法,用于测定硫酸西索米星氯化钠注射液的含量。新方法简便灵敏、重复性好,与抗生素微生物检定法相比,缩小了结果的置信区间,节省了检验时间。
1 仪器与试剂
1.1 试剂与药品
庚烷磺酸钠(HPLC级,日本东京化成工业株式会社),乙腈(HPLC级,Merck公司),超纯水(Milli-Q仪制备)。其他试剂均为市售,分析级。
硫酸西索米星氯化钠注射液(江苏亚邦生缘药业有限公司,批号:15062303、15090602、15101801);西索米星对照品(批号:130635-201301,含量56.0%),奈替米星对照品(批号:130355-200702)(中国食品药品检定研究院)。
1.2 仪 器
LC-30AD高效液相色谱仪(日本岛津公司)。
2 方法与结果
2.1 色谱条件
采用Aminoglycoside RP 18(4.6 mm×150 mm,3 μm)色谱柱;庚烷磺酸钠溶液(取庚烷磺酸钠6 g,加0.1 mol/L磷酸二氢钾溶液溶解并稀释至1 000 mL,用磷酸调节pH至1.5)-乙腈(77∶23)为流动相:流速1.0 mL/min;柱温:35 ℃;检测波长:205 nm;进样量:10 μL。
2.2 溶液配制
2.2.1 系统适用性溶液 取西索米星对照品和奈替米星对照品各适量,加水溶解并稀释制成每1 mL 中约含西索米星0.5 mg和奈替米星0.05 mg的混合溶液。
2.2.2 含量测定供试品溶液 精密量取硫酸西索米星氯化钠注射液适量,加水溶解并定量稀释制成每1 mL中约含西索米星0.5 mg的溶液,摇匀,作为供试品溶液。
2.2.3 含量测定对照品溶液 取硫酸西索米星对照品,用水溶解制成每1 mL中约含西索米星0.5 mg的溶液。
2.3 系统适用性试验
采用“2.2.1”项下溶液进行分离度和理论板数测定,结果显示西索米星主峰和前杂质分离良好,西索米星与奈替米星分离良好,西索米星峰理论板数为5 860,拖尾因子为0.82。本方法专属性良好。
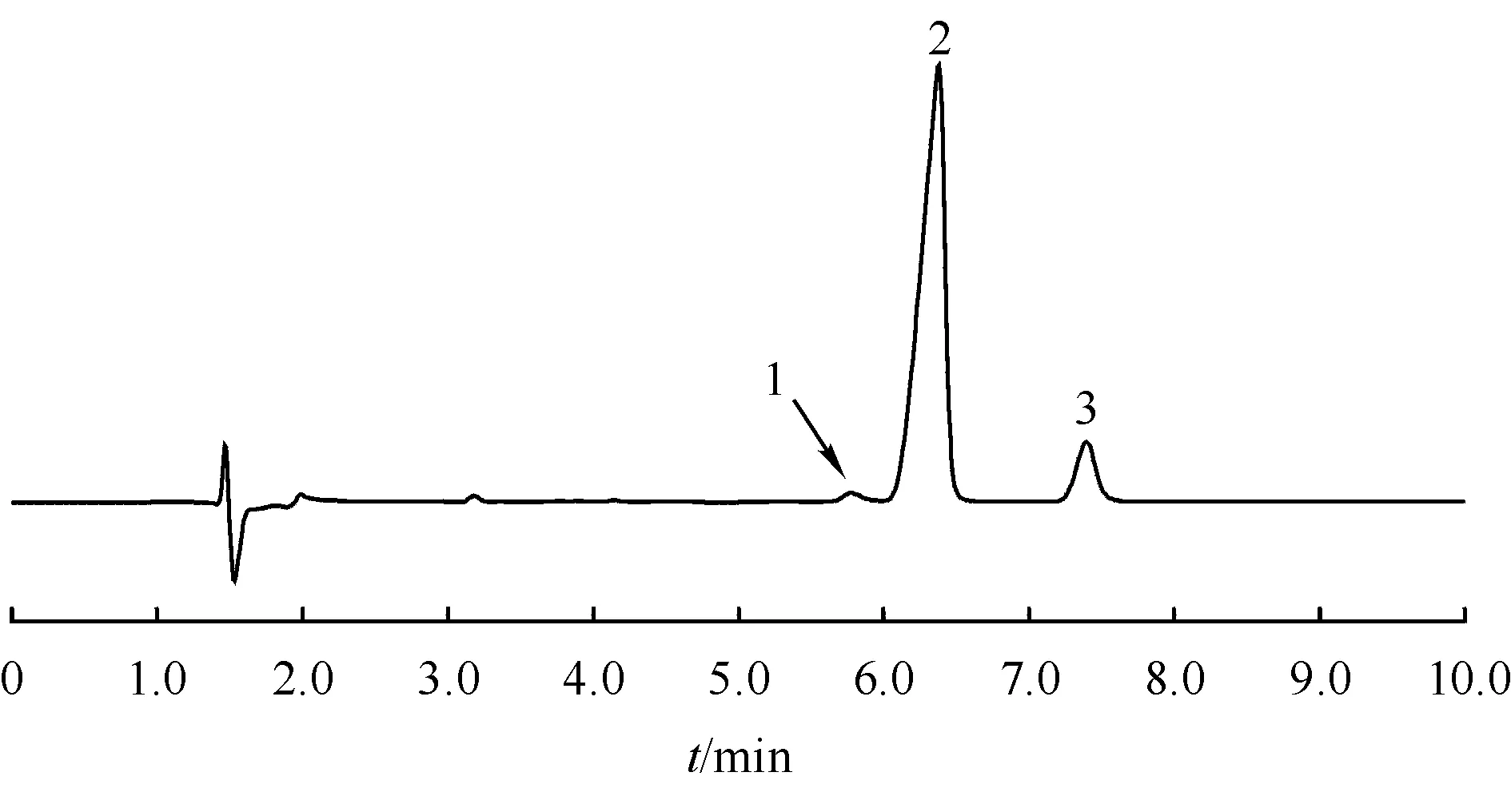
Figure1 Chromatogram of content determination (1:Unknown impurity;2:Sisomimy;3:Netilmicin)
2.4 破坏试验
取供试品适量,按“2.2.2”项下方法制成每1 mL 中约含西索米星1.0 mg的供试品溶液,分别经强酸(1 mol/L盐酸)1 mL沸水浴、强碱(1 mol/L氢氧化钠溶液)1 mL沸水浴、过氧化氢溶液1 mL、水浴90 ℃加热30 min和紫外光照(λ254 nm)24 h破坏处理,各液中和后分别用水稀释成每毫升中约含0.5 mg的供试品溶液;取空白溶剂同上进行破坏实验。各取10 μL进行HPLC分析。结果表明:在上述色谱系统下,西索米星色谱峰与各降解产物能良好分离,空白溶剂不干扰主成分的测定(西索米星主成分峰峰纯度阈值均>0.999 99,最小峰纯度指数均大于0)。破坏实验色谱图见图2。
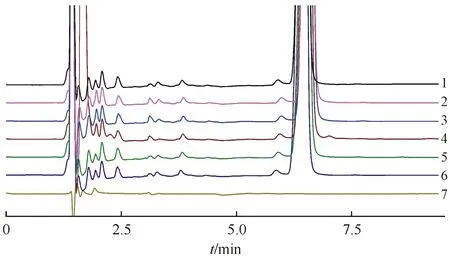
Figure2 Chromatogram of destruction test (1:Not destroyed;2:Acid destruction;3:Alkali destruction;4:Oxidative damage;5:Thermal damage;6:Light damage;7:Blank solvent)
2.5 检测限
将对照品溶液逐级稀释后进样10 μL,按S/N=3作为检测限,记录色谱图,定量限浓度约为0.06 μg/mL,相对西索米星供试品溶液浓度水平的0.000 1%。
2.6 定量限
将对照品溶液逐级稀释后进样10 μL,按S/N=10作为检测限,记录色谱图,定量限浓度约为0.2 μg/mL,相对西索米星供试品溶液浓度水平的0.000 4%。
2.7 线性范围
取西索米星对照品,加水溶解并稀释制成1.002 4、0.800 52、0.501 2、0.200 13、0.100 24、0.050 033、0.010 024 mg/mL的溶液,分别量取10 μL 注入液相色谱仪,记录峰面积,以浓度(X)和峰面积(Y)作线性回归,得回归方程为:Y=4.210 2×106X+9.107 0×103(r=0.999 9,n=7)。
西索米星浓度在0.010 024~1.002 4 mg/mL范围内与峰面积呈良好的线性关系。选择0.5 mg/mL 作为供试品测定浓度,即在供试品浓度的2%~200%范围内线性关系良好。
2.8 精密度
按“2.2.3”项下配制西索米星对照品溶液,连续进样6次,记录色谱图,计算主峰面积的RSD为0.09%,保留时间的RSD为0.11%,表明该方法精密度良好。
2.9 重复性
取批号为15062303的样品6份,按“2.2.2”项下条件处理得供试品溶液。分别取10 μL注入液相色谱仪测定含量,结果的RSD为0.76%,保留时间的RSD为0.19%,表明本方法重复性较好。
2.10 溶液稳定性
取批号为15062303的样品,按“2.2.2”项下条件处理得供试品溶液,分别置在0、2、4、6和8 h后测定,考察样品溶液的稳定性。结果主峰面积的RSD为0.33%,保留时间的RSD为0.13%,表明西索米星供试品溶液在8 h内稳定。
2.11 回收率试验
精密称取西索米星对照品,按处方配制样品溶液,用水稀释制成测定浓度的80%、100%、120%的溶液,按“2.1”项下条件分析,计算回收率,结果见表1。结果表明,方法的准确度良好。
2.12 方法的耐用性
采用3种品牌的耐酸色谱柱进样系统适用性溶液,考察方法的耐用性,结果前杂质峰与西索米星峰、西索米星与奈替米星峰均可以达到较为理想的分离,耐用性较好(见表2)。
Table1 Results of recovery rate test
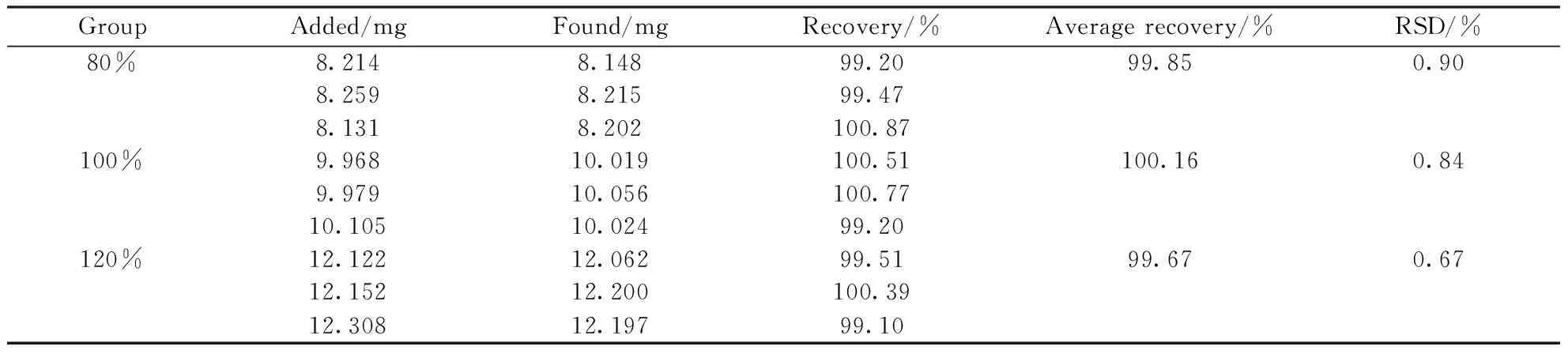
GroupAdded/mgFound/mgRecovery/%Average recovery/%RSD/%80%8.2148.14899.2099.850.908.2598.21599.478.1318.202100.87100%9.96810.019100.51100.160.849.97910.056100.7710.10510.02499.20120%12.12212.06299.5199.670.6712.15212.200100.3912.30812.19799.10
Table2 Separation effects under different columns
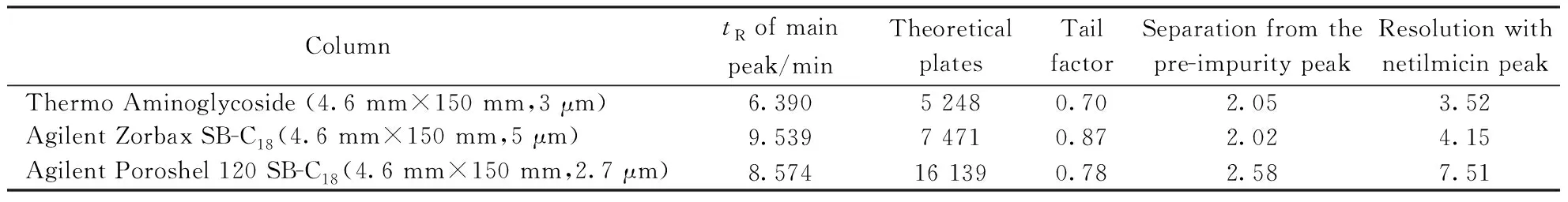
ColumntR of main peak/minTheoretical platesTail factorSeparation from the pre-impurity peakResolution with netilmicin peakThermo Aminoglycoside (4.6 mm×150 mm,3 μm)6.3905 2480.702.053.52Agilent Zorbax SB-C18 (4.6 mm×150 mm,5 μm)9.5397 4710.872.024.15Agilent Poroshel 120 SB-C18 (4.6 mm×150 mm,2.7 μm)8.57416 1390.782.587.51
采用Thermo Aminoglycoside (4.6 mm×150 mm,3 μm)色谱柱,略微改变“2.1”项下色谱条件的参数:柱温(32 ℃、35 ℃、38 ℃),流速(0.8 mL/min、1.0 mL/min、1.2 mL/min),流动相pH(pH 1.4、pH 1.5、pH 1.6)分别进样。系统适用性各峰分离良好,方法的耐用性良好。
2.13 含量测定
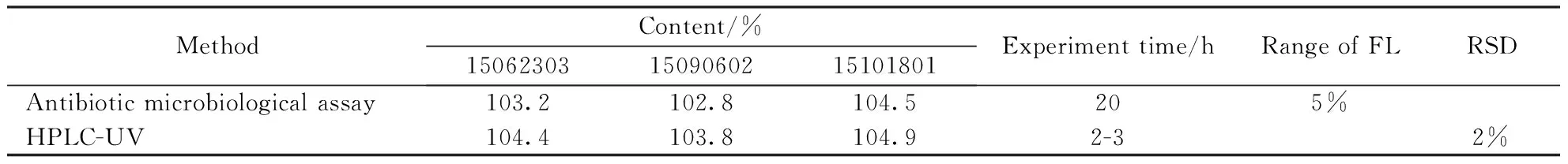
取硫酸西索米星氯化钠注射液,按“2.2.2”项下处理得供试品溶液,同法配制对照品溶液。分别精密量取供试品溶液和照品溶液各10 μL注入液相色谱仪,记录色谱图;按外标法计算西索米星的含量,并与抗生素微生物检定法进行比较。结果见表3。
Table3 Comparison of antibiotic microbial assay and HPLC methods
MethodContent/%150623031509060215101801Experiment time/hRange of FLRSDAntibiotic microbiological assay103.2102.8104.5205%HPLC-UV104.4103.8104.92-32%
结果显示,两种方法的含量测定结果无显著性差异。但从实验时间、成本以及误差范围综合考量,HPLC-UV方法有更大优势。
3 讨 论
3.1 方法的优化
《中华人民共和国药典》(2015年版)硫酸西索米星原料含量测定方法与USP40奈替米星含量测定方法色谱条件相同,均采用C18色谱柱,以庚烷磺酸钠溶液(取庚烷磺酸钠20.22 g,加0.07 mol/L的磷酸溶液溶解并稀释至1 000 mL)-乙腈(62∶38)为流动相。在该色谱条件下,西索米星峰难以与其前相邻杂质峰良好分离,且流动相离子对试剂浓度偏高,需较长的平衡时间。本文在中国药典方法基础上,降低流动相中庚烷磺酸钠的浓度(由2%至0.6%),用磷酸调节pH至1.5,优化后的色谱条件下西索米星峰与前相邻杂质峰以及西索米星峰与奈替米星峰均能达到良好的分离效果。
3.2 本文方法与抗生素微生物检定法的比较
抗生素微生物检定法可直观反映药品的抗菌活性,与临床应用的要求相符,能确定抗生素的医疗应用潜质,但该方法专属性较差,凡具有抗菌活性的物质都会干扰测定结果,且耗时长,对操作人员要求高且易产生诸多操作误差,重复性及精密度均较低。反相高效液相色谱法能使制剂中的组分与辅料有效分离,杂质及辅料对测定结果无干扰,灵敏度高,操作简单、快速,结果准确。本研究方法优化了中国药典所用HPLC方法的条件,提高了分离能力,大大降低了检验成本,同时满足了较高的精密度和准确度。本研究方法的测定结果与抗生素微生物检定法测定结果间没有显著差异,在实验时间、成本和置信区间等方面,HPLC-UV法具有更大的优势。
致谢:赵述强,张如梦同志收集整理相关资料并对研究工作给予帮助。
