运动对T2DM中骨钙素介导的能量代谢调控机制
2018-02-03李世昌方幸赵常红
徐 帅,李世昌,方幸,赵常红,
运动对T2DM中骨钙素介导的能量代谢调控机制
徐 帅1,2,李世昌1,2,方幸1,2,赵常红3,
1. 华东师范大学 体育与健康学院,上海 200241; 2. 华东师范大学 青少年健康评价与运动干预教育部重点实验室,上海 200241; 3. 西北民族大学 体育学院 甘肃 兰州 730124
s
骨钙素作为骨内分泌蛋白的角色逐渐受到重视,骨钙素能够作用于脂肪组织、胰腺和性腺以调节瘦素、脂联素、胰岛素和睾酮的分泌,进而维持机体能量代谢稳态;而T2DM存在脂质代谢紊乱、胰岛素分泌缺陷、β细胞功能障碍、性腺功能减退等现象,最终引起能量代谢异常的状况。不难看出,骨钙素和T2DM之间存在着相互性,而如何从骨分泌蛋白的角度去分析T2DM的能量代谢以及骨骼的内分泌能力未见报道。从骨钙素的内分泌功能进行分析,阐述了T2DM的研究现状、发病机制、运动干预等,为运动改善T2DM能量代谢提供新的视角。
T2DM;骨钙素;能量代谢;瘦素;胰岛素;运动
糖尿病会引起低骨量和骨折现象[28],而Ⅱ型糖尿病(Type 2 diabetes mellitus, T2DM)仍可保持骨密度不变,甚至可能略有升高[28,57]。但是,T2DM患者髋骨骨折的风险却会增加1.4~1.7倍,其骨密度却并未下降,究其原因,有研究认为,T2DM骨折不是因为骨量减少,而是由于骨质量恶化导致的骨结构改变[67]。目前在治疗T2DM过程中,有一些抗糖尿病药物没有对骨起到好的影响,服用后反而加重了原本就受损的骨结构。临床研究证实,骨构塑和骨重建是一种能量代谢的过程,骨骼与新陈代谢密切相关。当神经性厌食症患者摄食量减少,能量摄入降低,儿童出现总体性发育障碍,成年人出现低骨量[30,42]。近年来,骨内分泌系统(Bone endocrine system)作为能量代谢的新视角逐渐受到重视,骨骼可以通过分泌骨钙素(Osteocalcin, OCN)、成纤维细胞生长因子23(Fibroblast growth factor 23, FGF23)[3]和脂质蛋白2(Lipocalin 2, LCN2)[44]共同调节机体的内稳态。OCN在动物和人体内的代谢指数上升,会引起胰岛素敏感性、血清脂联素升高,胰岛素抵抗、血糖水平、脂肪重量和动脉硬化下降。运动作为调控T2DM代谢紊乱的干预手段,已经得到普遍认可。而运动如何通过骨分泌OCN调节T2DM的能量代谢过程,却鲜有研究报道。本文通过对OCN生物学机制加以阐释,同时在T2DM背景下分析OCN的调控机制,以期为运动改善T2DM骨代谢提供新的思路。
1 糖尿病引起的骨分泌异常
骨骼作为动态结缔组织,不断进行骨形成、骨构塑、骨重建等过程,从而保障骨骼内环境稳态。骨髓腔内的间充质干细胞(Mesenchymal stem cells, MSC)通常经前成骨细胞、不成熟成骨细胞、成熟成骨细胞,最终分化为骨细胞。临床研究发现,糖尿病骨量减少主要由于骨形成的减弱,而不是骨吸收的增加[6]。T2DM中骨转化过程的骨形成受损,导致成骨细胞的生成数量和功能下降。糖尿病患者会出现高血糖症、胰岛素功能损伤、血液流动性减弱等症状,引起骨基质中非酶糖化过程升高,导致晚期糖化终末产物(Advanced glycation end products, AGEs)的形成[61]。骨组织中AGEs急剧增加和堆积,直接抑制成骨细胞和骨细胞的分泌,引起OCN含量下降[8],有关研究证实,高糖和AGEs两者单独或共结合均可抑制成骨细胞,导致成骨功能紊乱,增强破骨活性[8,57],使得骨脆性提高。高血糖症也可直接抑制成骨细胞的表达和分泌[61];而成骨细胞的分化同样会受到糖尿病的抑制,在高糖中培养MSC,会导致成骨细胞/骨细胞的分化减弱,骨分泌OCN水平明显减少(图1)。
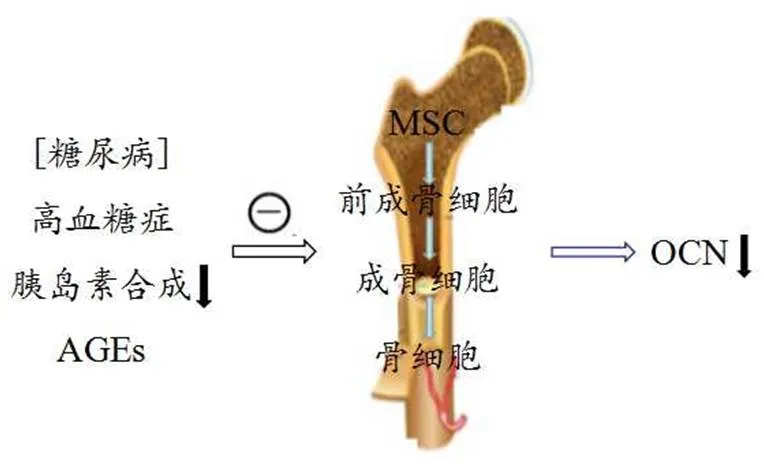
图1 糖尿病和骨分泌OCN联系示意图
Figure 1. The Relationship between Diabetes and Bone-generated OCN
2 骨钙素(OCN)
OCN是成骨细胞/骨细胞分泌的非胶原蛋白,由特异性谷氨酸(Glutamic acid, GLU)残基经羧化形成的γ-羧基谷氨酸(γ-carboxyglutamic acid, GLA)残基翻译、修饰后合成,也被称为骨γ-羧基谷氨酸蛋白(Bone γ-carboxyglutamic acid protein, Gla/BGP),分子量为5.6 KD,其中少部分OCN也由成牙质细胞分泌[51,69],小鼠OCN的羧化位点是GLU13、17、20;而人类OCN的羧化位点是GLU17、21、24,人类OCN基因位于1号染色体1q25-q31上,编码98个氨基酸[52]。成熟的OCN会通过分泌形式与羟磷灰石(Hydroxyapatite, HA)结合,储存在骨基质中[25]。羧化不全骨钙素(Uncarboxylated osteocalcin, uOCN)和羧化完全骨钙素(Carboxylated osteocalcin, cOCN)共同构成体内的总骨钙素(Total osteocalcin, tOCN)水平。骨基质中的OCN进入到骨吸收形成的高度酸性环境中(pH 4.5),由破骨细胞中的特异性T细胞免疫调节因子1(T-cell immune regulator 1, Tcirg1)编码产生的空泡质子泵对骨细胞外基质(Extracellular matrix, ECM)进行酸化,使OCN发生脱羧反应,最终形成有活性的uOCN[22,43]。uOCN可分泌入血,具有生物学功能,cOCN主要储存在骨骼中,无生物活性。uOCN通过调控糖代谢和脂肪组织的分泌能力,调控骨骼的新陈代谢[67]。在高脂膳食的野生型(WT)小鼠中,uOCN可改善脂肪细胞和胰岛素中β细胞分泌,以此达到预防肥胖、高血糖症等代谢性疾病的发生[19],而成骨细胞中基因敲除的OCN-/-小鼠会恶化这些过程[37]。下文将以OCN作为介导代谢调控中的核心因子,进行具体的分析和阐述(图2)。
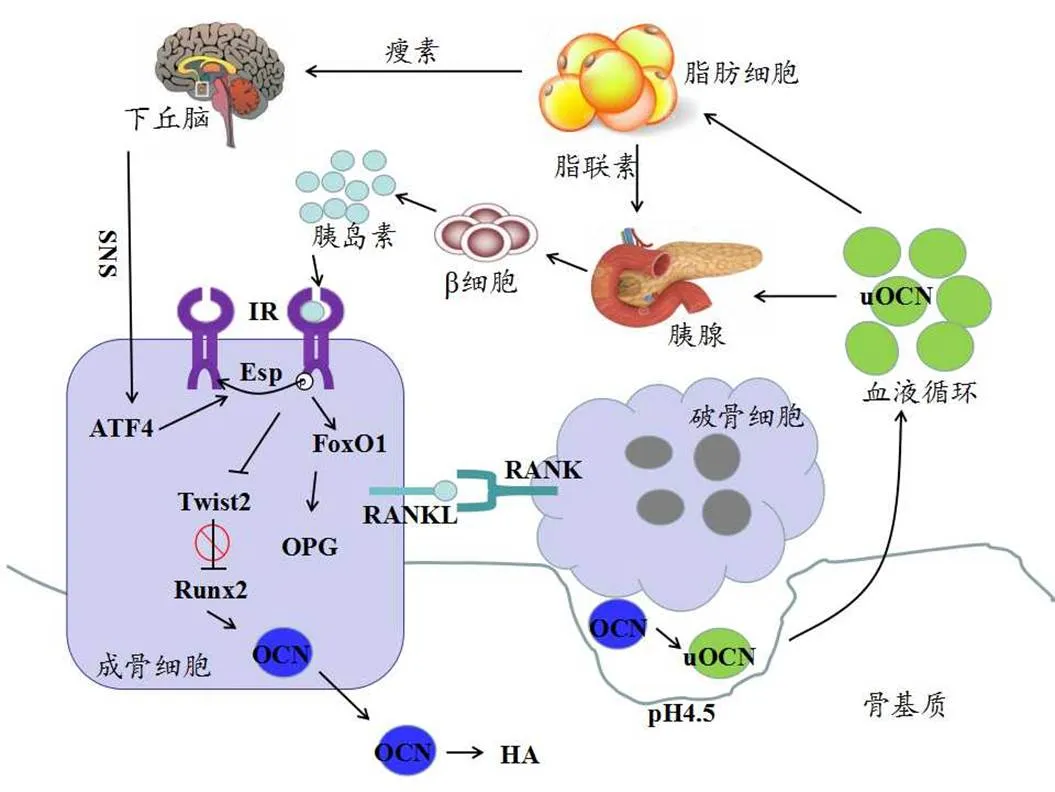
图2 OCN信号通路示意图[12,55]
Figure 2. OCN Signal Paths[12,55]
2.1 OCN介导的代谢调节
仅成骨细胞/骨细胞分泌的OCN进入血液循环,并介导瘦素和脂联素的能量代谢过程,而OCN-/-小鼠会引起脂肪堆积和糖代谢紊乱[17,37],Lee等[37]对调节血清uOCN水平的成骨细胞所富集基因进行筛选,确定出编码骨睾蛋白酪氨酸磷酸酶(Osteotesticular protein tyrosine phosphatase, OST-PTP)的胚胎干细胞磷酸酶(Embryonic stem cell phosphatase, Esp,又称Ptprv)基因。Esp是小鼠糖代谢的关键调节因子,只在成骨细胞和Sertoli细胞中进行表达,Esp结合胰岛素受体(Insulin receptor, IR),引起IR脱磷酸并致其失活,抑制胰岛素与IR结合,从而实现Esp对胰岛素信号通路的调节[26]。Esp-/-小鼠与OCN-/-小鼠代谢表型相反,Esp-/-小鼠表现出明显的血糖下降、脂肪量减少、β细胞增殖和胰岛素敏感性升高等生理变化[69],而将Esp-/-小鼠与OCN-/-小鼠杂交,可缓解Esp-/-小鼠的代谢紊乱情况。这些研究表明,成骨细胞中胰岛素信号是OCN活化和葡萄糖代谢的重要因素[20]。对特异性敲除成骨细胞中IR的小鼠(OB-ΔIR),可表现出明显的葡萄糖耐量、胰岛素敏感性和胰岛素分泌异常现象。
OCN可提高β细胞中Insulin1、Insulin2、CyclinD1、CyclinD2和CDK4表达,Ferron等[19]对WT小鼠糖代谢的研究中发现,当体外OCN增加后,可提高胰岛素表达和β细胞增殖;较高浓度OCN可增加白色脂肪和棕色脂肪组织中脂联素和过氧化物酶体增殖活化受体γ协同激活因子(Peroxisome proliferators activated receptor gamma coactivator-1, PPARGC1A)表达,PPARGC1A在体内能量代谢调节中扮演重要角色。活体皮下注射OCN会有效消除高脂饮食引起的代谢异常[69]。甚至对那些含有高脂肪量、高血糖的雌性个体,在其妊娠期内口服uOCN,其后代的能量代谢也可得到有效改善[32]。
2.2 OCN介导的瘦素调节
瘦素除限制饮食和能量消耗以外,在羊和人体实验中已经证实,瘦素可调节骨量[50],参与骨构塑和骨重建过程,是骨健康的重要标志之一[60],缺乏瘦素可观察到高骨量现象。uOCN作为骨和脂肪组织中的调节纽带,可直接影响二者的能量稳态。骨骼释放uOCN调节脂肪活性,引起瘦素分泌;瘦素间接抑制骨吸收,直接刺激骨形成[7,62]。在下丘脑调节过程中,瘦素与其受体(Leptin receptor, LepR)结合,抑制5-羟色胺(5-hydroxytryptamine, 5-HT)的合成,5-HT会与成骨细胞的Htr1b受体结合,引起成骨细胞中cAMP反应元件结合蛋白(cAMP responseelement binding protein, CREB)信号变化[46],瘦素与腹内侧下丘脑(VMH)的LepR结合,引起交感神经系统(Sympathetic nervous system, SNS)的活化并输出信号,再由成骨细胞中的β2-肾上腺素能受体(β2-adrenergic receptor, ADRβ2)接受。一方面,抑制成骨细胞增殖,引起Esp表达,并通过IR/FoxO1信号通路增加护骨素(Osteoprotegerin, OPG)的分泌,抑制OCN羧化;另一方面,增强骨吸收,促进OCN羧化,研究普遍认为,第1种反应效果更加强烈,而ADRB2缺失会导致小鼠骨量增加[2,20,30]。小鼠的LepR(Y985L)突变,会引起瘦素信号通路变化,出现骨质疏松症状[58]。研究发现,瘦素基因敲除(ob/ob)小鼠和LepR敲除(db/db)小鼠,在成骨细胞特异性未受到抑制时,未出现骨形成的增加[19,58]。而瘦素灌注到ob/ob小鼠第三脑室后,则导致骨量生成减弱,说明瘦素是先调节下丘脑,通过其内部分泌来改善骨代谢。瘦素经血脑屏障与下丘脑弓状核(Arc)中的LepR结合,进一步促进可卡因-苯丙胺调节转录肽(Cocaine and amphetamine regulated transcript, CART)基因表达,降低NF-Kβ受体活化因子配体(Receptor for activation of nuclear factor kappa β ligand, RANKL)在成骨细胞中的表达,OPG与RANKL比例不均会影响骨形成和骨吸收。小鼠的VMH或弓状神经元中LepR失活后进行普通膳食喂养,发现并未影响其饮食和骨量生长,中缝核分泌5-HT与瘦素也存在直接的交互作用,共同抑制骨骼的自然生长[2,30]。
有趣的是,除了对食欲和骨量的调节,瘦素还通过直接或间接的方式调控胰腺分泌。瘦素对胰岛素分泌的间接作用取决于SNS对成骨细胞的调节,瘦素通过刺激SNS,活化激活转录因子4(Activating transcription factor 4, ATF4),改善成骨细胞中的Esp表达,引起uOCN含量变化[20,27]。OCN可促进单胺神经递质的合成,抑制γ氨基丁酸合成,并改变多巴胺和去甲肾上腺素水平。uOCN还会穿过血脑屏障,促进脑干、中脑和海马中神经递质的合成,改善学习和记忆能力[69]。
2.3 OCN与胰岛素的相互调节
T2DM存在胰岛素抵抗和β细胞功能障碍,抑制成骨细胞中uOCN的循环水平[9]。OST-PTP通过促进酪氨酸磷酸化,以及IR失活来减弱成骨细胞中的胰岛素信号。胰岛素信号传导通过OST-PTP的磷酸化水平影响糖代谢。Fulzele等[22]在讨论成骨细胞中胰岛素活性时,发现OCN对糖代谢的调节作用。研究指出,OB-ΔIR小鼠与Karsenty等[30]探讨的OCN-/-小鼠代谢表型相似。对OB-ΔIR小鼠进行2周的uOCN干预,即可提高胰岛素敏感性[43]。Esp-/-小鼠与OB-ΔIR小鼠相比,出现低血糖症、高胰岛素血症、β细胞增殖和胰岛素敏感性增强现象[43],而OB-ΔIR小鼠体内脂肪量堆积,出现血清胰岛素水平、胰岛素敏感性、糖耐量下降等现象。从OCN与胰岛素突变小鼠给药改善糖代谢的报道来看,确实存在骨-胰腺轴(Bone pancreas axis)的内分泌回路[69]。Ferron等[21]已经证实胰腺和骨骼的相互调节作用,成骨细胞中的胰岛素信号可提高OCN和糖代谢水平,增加的uOCN又会提高胰岛素分泌量和胰岛素敏感性。而用破骨细胞增益和功能丧失模型[35]发现,OPG-/-小鼠可提高破骨细胞活性和葡萄糖耐量,uOCN含量在OPG-/-小鼠中有所升高,而小鼠中破骨细胞的消融会导致葡萄糖耐量降低,表明,破骨细胞通过对OCN脱羧调节机体代谢。
IR磷酸化可引起FoxO1和Twist2变化[22]。成骨细胞中FoxO1缺失可降低Esp表达,增加OCN合成,提高胰岛素生成和胰岛素敏感性[5]。成骨细胞转录因子缺陷(FoxO1flox)小鼠可增强β细胞增殖、胰岛素分泌和胰岛素敏感性,使OCN分泌量和生物活性升高[34,69]。特异性敲除成骨细胞FoxO1(FoxO1osb-/-)小鼠也会引起OPG表达下降、破骨细胞生成增加、葡萄糖耐量和胰岛素敏感性的提高,引起血清uOCN含量增加。由FoxO1直接结合Esp启动子,引起Esp的表达量下降,从而调节OCN表达[53];ATF4也可直接诱导成骨细胞中Esp表达。FoxO1还会与ATF4共同调控OCN的分泌,协同诱导成骨细胞中Esp表达,共同改善葡萄糖耐受水平[34]。
研究表明,OB-ΔIR小鼠会引起Twist2表达,出现较低OCN循环[11,22],Twist2的下游调控因子——核心结合因子2(Runt related transcription factor 2, Runx2)受到抑制,Runx2与OCN启动子的结合显著降低,OCN含量随之减少。在后期成长中,OB-ΔIR小鼠出现典型的外围性肥胖和高血糖症,当注入uOCN后,代谢性异常情况则会得到明显改善,进而提高胰岛素敏感性和胰岛素分泌量,也就是骨-胰腺内分泌循环的调控作用[22]。胰岛素渗透测定法显示,细胞溶质Ca2 +水平增加,uOCN可提高胰岛素分泌水平,uOCN通过刺激小肠内胰高血糖素样肽-1(Glucagon-like peptide-1, GLP-1),间接刺激胰岛素分泌;口服GLP-1也可刺激体内uOCN的增加,同时刺激胰岛素分泌[43]。这些研究均表明,成骨细胞产生的OCN通过直接影响胰岛的生物功能,即胰岛素合成和分泌来控制机体的葡萄糖代谢。
2.4 OCN对生殖功能的调节
男性T2DM患者与性腺机能减退密切相关[16]。与健康组相比,T2DM小鼠和患者睾酮水平均较低,T2DM患者存在睾丸损伤和睾酮分泌不足[13]。睾酮含量与OCN有着直接关系,成骨细胞产生OCN可提高睾丸分泌能力,OCN-/-小鼠可导致生殖能力下降[30]。OCN-/-小鼠表现为睾酮含量下降、精液减少、生殖器官萎缩;体内注射OCN后,小鼠的生殖能力又会得到有效改善。有趣的是,Esp-/-小鼠在提高成骨细胞的胰岛素信号时,也会提高雄性生殖水平[21],说明,胰岛素可通过刺激骨转换和骨细胞中OCN活性来提高雄性生殖功能。胰腺-骨-性腺轴(pancreas-bone-testis axis)作为调节雄性生殖功能的关键轴之一,已得到广泛关注[45]。GPRC6A属于G蛋白偶联受体C家族6组A(G protein-coupled receptor family class C group 6 subtype A),存储于睾丸的Leydig细胞中,也是目前唯一被确定的OCN受体[14]。GPRC6A-/-小鼠与WT小鼠相比具有更多的白色脂肪、肝脂肪变性、葡萄糖不耐受、胰岛素抵抗等生物学特性,以及睾酮低水平等差异[47]。GPRC6A-/-小鼠与OCN-/-小鼠表现出相似的表征:脂肪堆积、高血糖症、葡萄糖耐受不良、胰岛素抵抗和雄性生殖功能减退[47]。
3 运动干预对T2DM中OCN分泌影响
T2DM患者每周进行150 min的中等强度有氧运动,可显著改善T2DM中血糖和胰岛素抵抗,但对提高T2DM的骨质疏松效果还远未达到。而抗阻运动和耐力训练等可对骨骼产生直接刺激,上调OPG水平,显著提高OPG/RANKL的比例,明显增加骨密度,提高骨质量[49,59,63]。研究还指出,T2DM患者多采用抗阻运动或联合运动(有氧运动和抗阻运动)效果更佳[18]。T2DM对MSC分化具有显著的导向作用[54]。T2DM患者骨髓腔内MSC向脂肪细胞分化增强,成骨分化随之减弱,骨密度下降。临床采用噻唑烷二酮类药物(TZDs)对T2DM患者进行治疗,但会引起MSC优先向脂肪细胞分化,成骨细胞分化减弱,增大骨脆性[10]。运动可引导T2DM中MSC的分化途径,提高T2DM中成骨细胞/骨细胞的合成,增加OCN的含量[33],提高运动者血清uOCN水平,特别是对T2DM肥胖患者,有氧运动能够增加血清uOCN水平,从而调节葡萄糖水平[15]。对4周龄C57BL/6雄性小鼠进行耐力训练,可显著增强MSC成骨分化[68]。在小鼠爬梯运动模型中发现,28天爬梯训练小鼠,MSC的成骨潜能显著增加,骨髓中脂肪细胞数量下降,成骨细胞数量升高[41],进而增加OCN的分泌量。
3.1 运动对T2DM中OCN介导能量调节
肥胖型T2DM男性进行急性有氧运动和力量运动后,血糖水平可有效下降,并发现OCN与血糖变化有直接关联。急性有氧运动后,OCN羧化水平提高,血清uOCN含量上升,促进胰腺分泌胰岛素,或提高胰岛素敏感性,进而降低T2DM的血糖水平;力量运动虽会调节糖代谢,但uOCN的含量只得到少量提升,这可能是骨骼肌代谢引起的部分葡萄糖代谢[38,39]。有研究对39名肥胖男性进行8周的运动干预后,发现uOCN含量升高,体脂质量和血清瘦素水平明显降低,胰岛素抵抗得到改善;通过限制卡路里摄入和运动干预后,明显增加血清uOCN含量,由于uOCN与脂肪细胞之间的相关性,研究认为,脂肪量是预测uOCN变化的最佳参数[34]。Gravenstein的SEqM模型说明,瘦素与脂肪量密切相关,同时,瘦素还会负性调控ATF4和Esp调控的FoxO1水平[23,34]。在小鼠和人类的耐力运动中,uOCN水平也可显著增加[24]。对T2DM型OLETF大鼠的研究发现,二甲双胍治疗组以及二甲双胍和耐力运动结合组中,瘦素分泌下降,而在单独的耐力运动组中瘦素并未出现下降;血清参数显示,耐力运动中血糖水平、胰岛素抵抗、HbA1c显著下降[29]。耐力运动对于治疗T2DM症状优于药物治疗和药物+运动共结合的效果。定期规律运动可保持β细胞质量,进行适当的剧烈运动(>80%的最大运动能力)可提高儿茶酚胺和皮质醇释放,促进早期恢复期间的胰岛素反应,以抵消瞬时高血糖[9],进而降低AGEs的含量,削弱对成骨分化的抑制效果,从而提高OCN分泌量,保证血清uOCN水平。
对于OCN介导的瘦素调控通路,研究认为,瘦素是通过2种不同的神经通路作用于成骨细胞:1)通过SNS作用于成骨细胞上的受体,诱导RANKL表达,有利于破骨细胞的分化;2)通过下丘脑的CART基因抑制RANKL表达[36]。运动还会使得RANKL表达减弱,抑制破骨细胞分化,提高骨质量。阻塞RANKL信号通路,可有效预防T2DM发生[65]。对71名老年女性的运动干预发现,8个月的抗阻和有氧运动,能够改善骨密度,但对OPG/RANKL水平影响不大,其可能是由于老年女性的运动强度较低,达不到刺激水平,使得瘦素分泌对RANKL的调节维持在稳定的平衡状态[40]。对T2DM患者进行抗阻运动,瘦素分泌减弱,脂联素水平反而提高,改善了胰岛素敏感性,进而推测,T2DM患者中,瘦素会抑制脂联素发挥胰岛素的增敏作用[1]。瘦素基因移入型T2DM小鼠,摄食量、饮水量、总胆固醇和血糖水平显著下降[66]。跑轮运动能明显提高大鼠下丘脑的LepR蛋白表达水平,运动还可以提高糖尿病大鼠的大脑皮质和瘦素信号[4]。
3.2 运动对OCN介导的生殖调节
流行病学显示,睾酮水平与胰岛素敏感性存在直接相关性,低睾酮会增加罹患T2DM的可能性,前列腺癌患者获T2DM的风险尤为显著。较低的睾酮和性激素结合球蛋白(Sex hormone-binding globulin, SHBG)结合,会提高代谢综合征的发生率,而睾酮水平升高后,可逆转性腺机能减退引起的T2DM所导致不良状况[56]。睾酮作为雄激素中最主要的代谢性激素之一,其生物学作用包括促进肌肉生长[64]和维持骨骼质量[48]。运动提高成骨细胞OCN分泌量,引起血清uOCN与GPRC6A结合,活化cAMP/PKA/CREB通路,引起CREB的磷酸化,提高StAR、Cyp11ɑ、3β-HSD和Cyp17等关键酶活性,最终引起睾酮含量的升高[31]。男性睾酮水平在青春期前受到运动的调控作用不大,而在青春期后期,抗阻运动会引起睾酮的急剧增加,男性超过35~40岁后,血液循环睾酮浓度会以每年1%~3%水平下降,抗阻运动可有效缓解血睾酮下降的趋势[64],推测是由于运动提高OCN含量,引起的睾酮水平升高。
综上所述,OCN的生物活性在基因表达、能量代谢和生殖能力上都具有显著的调节功能。在运动改善T2DM的情况下,还会引起骨内分泌的改善,提高OCN的分泌和血清uOCN水平;同时,运动还通过调节脂肪组织、下丘脑、胰腺和性腺等分泌,共同构成一个多器官、多回路、共循环的调节系统,从而做到相互影响,共同改善的效果(图3)。但是,对于运动的方式和方法,是如何改善T2DM中OCN信号通路的探讨尚未开展,值得进一步的研究。

图3 运动通过骨内分泌对多器官的作用示意图
Figure 3. The Effect of Exercise on Multiple organs by Endocrine
注:T2DM本身对骨骼、脂肪组织、下丘脑、胰腺和性腺等存在着负性调控作用。图中实线代表运动对各组织器官的作用以及运动改善T2DM的效果;虚线代表运动通过血清uOCN对各组织器官的调节方式,且各组织器官也会对骨骼产生相应的反馈。
4 小结
OCN作为骨骼分泌的能量代谢特异性调控因子,已经受到广泛关注。OCN对脂肪细胞、下丘脑、胰腺和性腺等都具有重要的调控作用,其介导分泌的瘦素、脂联素、胰岛素和睾酮在能量代谢,以及机体的生殖能力中均具有重要的调节功能。而T2DM存在着能量代谢紊乱、骨质量下降、高血糖症和性腺功能下降等症状,与OCN介导方式和作用有许多共同之处。作为OCN的活性形式,uOCN通过血液循环方式,再作用于各个靶器官,引起瘦素、脂联素、胰岛素和睾酮的进一步调节,uOCN的受体除GPRC6A外,其他靶器官的对接受体尚不清楚,仍然需要进一步的验证。国内、外对于T2DM的骨分泌探索研究相对较少,且多数停留在血清OCN和uOCN的分析上,关于运动改善T2DM中OCN的信号机制,相关研究未见报道。其中,研究的难度多集中于靶向调控的分析和界定,尤其是T2DM自身的内分泌破坏和骨内分泌的双重干扰,这也逐渐成为研究的热点和难点。运动作为有效的干预手段之一,对改善T2DM存在多方面的影响,其运动本身就会对T2DM的脂肪细胞、下丘脑、胰腺和性腺具有良好的调节作用,通过OCN信号通路来解决T2DM的生理、病理的相关机制,虽然得到普遍共识,但其中具体的骨内分泌调控机制,以及临床意义仍然需要进一步的研究。
[1] 黄彩华, 陈俊钦, 林建新, 等. 运动改善2型糖尿病胰岛素抵抗与血清脂联素和瘦素及其交互作用[J]. 中国体育科技, 2011(04): 100-105.
[2] 王春玉, 龙丰云, 陈香, 等. 骨钙素介导的骨内分泌系统[J]. 中华骨质疏松和骨矿盐疾病杂志, 2013,(3): 196-202.
[3] 徐帅, 李世昌, 方幸. 运动与骨内分泌系统研究进展[J],体育学刊, 2017,37(3): 139-144.
[4] 薛香莉, 刘微娜, 漆正堂, 等. 脂肪细胞因子与运动的抗抑郁作用[J]. 体育科学, 2016,36(11):66-74.
[5] 杨娟, 佘美华. FoxO1在2型糖尿病中的作用及其研究进展[J]. 中南医学科学杂志, 2017,(3):307-311.
[6] ACHEMLAL L, TELLAL S, RKIOUAK F,. Bone metabolism in male patients with type 2diabetes[J]. Clin Rheumatol, 2005, 24(5): 493-496.
[7] BALDOCK P. Reciprocal regulation of bone and energy metabolism[J]. Horm Res Paediatr, 2011,76(Suppl): 7-11.
[8] BASTA G, SCHMIDT A M, DE CATERINA R. Advanced glycation end products and vascularinflammation: implications for accelerated atherosclerosis in diabetes[J]. Cardiovasc Res, 2004, 63(4):582-592.
[9] BEAUDRY J L, RIDDELL M C. Effects of glucocorticoids and exercise on pancreatic beta-cellfunction and diabetes developm-ent[J]. Diabetes Metab Res Rev, 2012, 28(7): 560-573.
[10] BENVENUTI S, CELLAI I, LUCIANI P,. Rosiglitazone sti-mulates adipogenesis anddecreases osteoblastogenesis in human mesenchymal stem cells[J]. J Endocrinol Invest, 2007, 30(9):C26-C30.
[11] BOOTH S L, CENTI A J, GUNDBERG C. Bone as an endocrine organ relevant to diabetes[J].Curr Diab Rep, 2014, 14(12): 1-8.
[12] BOOTH S L, CENTI A, SMITH S R,. The role of osteocalc-in in human glucose metabolism: Marker or mediator?[J]. Nat Rev Endocrinol, 2013, 9(1): 43-55.
[13] BOYANOV M A, BONEVA Z, CHRISTOV V G. Testosterone supplementation in men with type2 diabetes, visceral obesity and partial androgen deficiency[J]. Aging Male, 2003, 6(1): 1-7.
[14] CLEMMENSEN C, SMAJILOVIC S, WELLENDORPH P,. The GPCR, class C, group 6,subtype A (GPRC6A) receptor: From cloning to physiological function[J]. Br J Pharmacol, 2014,171(5): 1129-1141.
[15] CONFAVREUX C B, LEVINE R L, KARSENTY G. A paradigm of integrative physiology, thecrosstalk between bone and energy metabolisms[J]. Mol Cell Endocrinol, 2009, 310(1-2): 21-29.
[16] CORONA G, MONAMI M, RASTRELLI G,. Type 2 diabetes mellitus and testosterone: A meta-analysis study[J]. Int J Androl, 2011, 34(6 Pt 1): 528-540.
[17] DUCY P, DESBOIS C, BOYCE B,. Increased bone formation in osteocalcin-deficientmice[J]. Nature, 1996, 382(6590): 448-452.
[18] DUNSTAN D. Diabetes: Exercise and T2DM-move muscles more often![J]. Nat Rev Endocrinol, 2011, 7(4): 189-190.
[19] FERRON M, HINOI E, KARSENTY G,. Osteocalcin differentially regulates beta cell andadipocyte gene expression and affects the development of metabolic diseases in wild-type mice[J].Proc Natl Acad Sci U S A, 2008, 105(13): 5266-5270.
[20] FERRON M, LACOMBE J. Regulation of energy metabolism by the skeleton: Osteocalcin andbeyond[J]. Arch Biochem Biophys, 2014, 561: 137-146.
[21] FERRON M, WEI J, YOSHIZAWA T,. Insulin signaling in osteoblasts integrates boneremodeling and energy metabolism[J]. Cell, 2010, 142(2): 296-308.
[22] FULZELE K, RIDDLE R C, DIGIROLAMO D J,. Insulin receptor signaling in osteoblastsregulates postnatal bone acquisit-ion and body composition[J]. Cell, 2010, 142(2): 309-319.
[23] GRAVENSTEIN K S, NAPORA J K, SHORT R G,. Cross-sectional evidence of a signalingpathway from bone homeostasis to glucose metabolism[J]. J Clin Endocrinol Metab, 2011, 96(6):E884-E890.
[24] GREENHILL C. Exercise: Osteocalcin in the adaptation to exerc-ise[J]. Nat Rev Endocrinol, 2016,12(8): 434.
[25] GUNDBERG C M, CLOUGH M E. The osteocalcin propeptide is not secreted in vivo or invitro[J]. J Bone Miner Res, 1992, 7(1): 73-80.
[26] GUNTUR A R, ROSEN C J. Bone as an endocrine organ[J]. Endocr Pract, 2012, 18(5): 758-762.
[27] HINOI E, GAO N, JUNG D Y,. The sympathetic tone medi-ates leptin's inhibition of insulinsecretion by modulating osteocal-cin bioactivity[J]. J Cell Biol, 2008, 183(7): 1235-1242.
[28] ISAIA G C, ARDISSONE P, DI STEFANO M,. Bone meta-bolism in type 2 diabetesmellitus[J]. Acta Diabetol, 1999, 36(1-2): 35-38.
[29] JENKINS N T, PADILLA J, ARCE-ESQUIVEL A A,. Effe-cts of endurance exercise training,metformin, and their combina-tion on adipose tissue leptin and IL-10 secretion in OLETF rats[J]. J ApplPhysiol (1985), 2012, 113(12): 1873-1883.
[30] KARSENTY G, FERRON M. The contribution of bone to whole-organism physiology[J]. Nature, 2012, 481(7381): 314-320.
[31] KARSENTY G, OURY F. Regulation of male fertility by the bone-derived hormoneosteocalcin[J]. Mol Cell Endocrinol, 2014, 382(1): 521-526.
[32] KAWAKUBO-YASUKOCHI T, KONDO A, MIZOKAMI A,. Maternal oral administrationof osteocalcin protects offspring from metabolic impairment in adulthood[J]. Obesity (Silver Spri-ng), 2016, 24(4): 895-907.
[33] KIM Y S, NAM J S, YEO D W,. The effects of aerobic exercise training on serumosteocalcin, adipocytokines and insulin resistance on obese young males[J]. Clin Endocrinol (Oxf), 2015, 82(5): 686-694.
[34] KODE A, MOSIALOU I, SILVA B C,. FoxO1 protein cooperates with ATF4 protein inosteoblasts to control glucose homeostasis[J]. J Biol Chem, 2012, 287(12): 8757-8768.
[35] LACOMBE J, KARSENTY G, FERRON M. In vivo analysis of the contribution of boneresorption to the control of glucose metabolism in mice[J]. Mol Metab, 2013, 2(4): 498-504.
[36] LEE N K, KARSENTY G. Reciprocal regulation of bone and energy metabolism[J]. JMusculoskelet Neuronal Interact, 2008, 8(4): 351.
[37] LEE N K, SOWA H, HINOI E,. Endocrine regulation of energy metabolism by theskeleton[J]. Cell, 2007, 130(3): 456-469.
[38] LEVINGER I, JERUMS G, STEPTO N K,. The effect of acute exercise onundercarboxylated osteocalcin and insulin sensitivity in obese men[J]. J Bone Miner Res, 2014, 29(12):2571-2576.
[39] LEVINGER I, ZEBAZE R, JERUMS G,. The effect of acute exercise on undercarboxylatedosteocalcin in obese men[J]. Osteo-poros Int, 2011, 22(5): 1621-1626.
[40] MARQUES E A, WANDERLEY F, MACHADO L,. Effects of resistance and aerobicexercise on physical function, bone mineral density, OPG and RANKL in older women[J]. ExpGero-ntol, 2011, 46(7): 524-532.
[41] MENUKI K, MORI T, SAKAI A,. Climbing exercise enha-nces osteoblast differentiation andinhibits adipogenic differentia-tion with high expression of PTH/PTHrP receptor in bone marrowcells[J]. Bone, 2008, 43(3): 613-620.
[42] MISRA M, KLIBANSKI A. The neuroendocrine basis of anorexia nervosa and its impact on bonemetabolism[J]. Neuroendocrinology, 2011, 93(2): 65-73.
[43] MIZOKAMI A, KAWAKUBO-YASUKOCHI T, HIRATA M. Osteocalcin and its endocrinefunctions[J]. Biochem Pharmacol, 2017, 132: 1-8.
[44] MOSIALOU I, SHIKHEL S, LIU J M,MC4R-dependent suppression of appetite bybone-derived lipocalin 2[J]. Nature, 2017, 543(7645): 385-390.
[45] OURY F, FERRON M, HUIZHEN W,. Osteocalcin regulates murine and human fertilitythrough a pancreas-bone-testis axis[J]. J Clin Invest, 2013, 123(6): 2421-2433.
[46] OURY F, YADAV V K, WANG Y,. CREB mediates brain serotonin regulation of bone massthrough its expression in ventromedial hypothalamic neurons[J]. Genes Dev, 2010, 24(20): 2330-2342.
[47] PI M, CHEN L, HUANG M Z,. GPRC6A null mice exhibit osteopenia, feminization andmetabolic syndrome[J]. PLoS One, 2008, 3(12): e3858.
[48] PI M, QUARLES L D. Multiligand specificity and wide tissue ex-pression of GPRC6A revealsnew endocrine networks[J]. Endocr-inology, 2012, 153(5): 2062-2069.
[49] PICHLER K, LORETO C, LEONARDI R,. RANKL is downregulated in bone cells byphysical activity (treadmill and vibration stimulation training) in rat with glucocorticoid-inducedosteoporosis[J]. Histol Histopathol, 2013, 28(9): 1185-1196.
[50] POGODA P, EGERMANN M, SCHNELL J C,. Leptin inhibits bone formation not only inrodents, but also in sheep[J]. J Bone Miner Res, 2006, 21(10): 1591-1599.
[51] PRICE P A, POSER J W, RAMAN N. Primary structure of the gamma-carboxyglutamicacid-containing protein from bovine bone[J]. Proc Natl Acad Sci U S A, 1976, 73(10): 3374-3375.
[52] PUCHACZ E, LIAN J B, STEIN G S,. Chromosomal localization of the human osteocalcingene[J]. Endocrinology, 1989, 124(5): 2648-2650.
[53] RACHED M T, KODE A, SILVA B C,. FoxO1 expression in osteoblasts regulates glucosehomeostasis through regulation of osteocalcin in mice[J]. J Clin Invest, 2010, 120(1): 357-368.
[54] RIBOT J, CALIAPEROUMAL G, PAQUET J,. Type 2 diabetes alters mesenchymal stemcell secretome composition and angiogenic properties[J]. J Cell Mol Med, 2017, 21(2): 349-363.
[55] ROSEN C J, MOTYL K J. No bones about it: insulin modulates skeletal remodeling[J]. Cell, 2010,142(2): 198-200.
[56] SAAD F, GOOREN L J. The role of testosterone in the etiology and treatment of obesity, themetabolic syndrome, and diabetes mellitus type 2[J]. J Obes, 2011, 1-10.
[57] SCHWARTZ A V. Diabetes Mellitus: Does it Affect Bone?[J]. Calcif Tissue Int, 2003, 73(6):515-519.
[58] SHI Y, YADAV V K, SUDA N,. Dissociation of the neuronal regulation of bone mass andenergy metabolism by leptin in vivo[J]. Proc Natl Acad Sci U S A, 2008, 105(51): 20529-20533.
[59] SPELLMAN C W. Aggressively managing type 2 diabetes mellitus, hyperlipidemia, and boneloss[J]. J Am Osteopath Assoc, 2008, 108(5 Suppl 3): S20-S27.
[60] TANNA N, PATEL K, MOORE A E,. The relationship between circulating adiponectin,leptin and vaspin with bone mineral density (BMD), arterial calcification and stiffness: A cross-sectional study in post-menopausal women[J]. J Endocrinol Inve-st, 2017,1-9.
[61] TERADA M, INABA M, YANO Y,. Growth-inhibitory effect of a high glucoseconcentration on osteoblast-like cells[J]. Bone, 1998, 22(1): 17-23.
[62] VASILKOVA O, MOKHORT T, SHARSHAKOVA T,. Leptin is an independentdeterminant of bone mineral density in men with type 2 diabetes mellitus[J]. Acta Diabetol, 2011,48(4): 291-295.
[63] VINCENT K R, BRAITH R W. Resistance exercise and bone turnover in elderly men andwomen[J]. Med Sci Sports Exerc, 2002, 34(1): 17-23.
[64] VINGREN J L, KRAEMER W J, RATAMESS N A,. Testo-sterone physiology in resistanceexercise and training: the up-stream regulatory elements[J]. Sports Med, 2010, 40(12): 1037-1053.
[65] WILSON C. Diabetes: Blocking RANKL signalling might prevent T2DM[J]. Nat Rev Endocrinol, 2013, 9(4): 188.
[66] XIANG L, LI J, WANG Q,. Leptin gene transfer improves symptoms of type 2 diabetic mice by regulating leptin signaling pathway and insulin resistance of peripheral tissues[J]. HumGene Ther, 2017, PMID: 28622065.
[67] YAMAGUCHI T, SUGIMOTO T. Bone metabolism and fracture risk in type 2 diabetes mellitus[J]. Endocr J, 2011, 58(8): 613-624.
[68] YUAN Y, CHEN X, ZHANG L,. The roles of exercise in bone remodeling and in preventionand treatment of osteoporosis[J]. Prog Biophys Mol Biol, 2016, 122(2): 122-130.
[69] ZOCH M L, CLEMENS T L, RIDDLE R C. New insights into the biology of osteocalcin[J]. Bone, 2016, 82: 42-49.
Mechanism of Exercise Regulates Energy Metabolism by Osteocalcin in T2DM
XU Shuai1,2,LI Shi-chang1,2,FANG Xing1,2,ZHAO Chang-hong3
1.East China Normal University, Shanghai 200241, China; 2. East China Normal University, Shanghai 200241, China; 3. Northwest University for Nationalities, Lanzhou 730124, China.
As a bone endocrine protein, Osteocalcin(OCN) obtain much larger attention over time. OCN regulates the secretion of leptin, adiponectin, insulin and testosterone through the adipose tissue, pancreas and gonadal, consequently maintains the steady state for body energy metabolism. Remarkably, type 2 diabetes mellitus (T2DM) tends to be caused by adipocyte disorder, insulin secretion deficiency, β cells dysfunction as well as reproduction decline, which finally contributes to abnormal energy metabolism. As a result, there is an interrelation between osteocalcin and T2DM. Unfortunately, few research involve energy metabolism of T2DM as well as bone differentiation in the perspective of bone endocrine protein. In this paper, the endocrine capacity of OCN is analyzed. Furthermore, the landscape, pathogenesis as well as exercise intervention for T2DM are proposed, perspective a new potential mechanism of exercise improving energy metabolism in T2DM.
G804.7
A
1002-9826(2018)01-0129-07
10.16470/j.csst.201801018
2017-05-23;
2017-12-20
国家社会科学基金“十三五”规划课题(BLA170225);华东师范大学研究生科研创新实践项目(ykc17049)。
徐帅,男,在读硕士研究生,主要研究方向为运动与骨分子生物学, E-mail:xus1992@126.com。
