琚湾酸浆面浆水细菌多样性评价
2018-01-31周书楠席修璞叶万海张振东
周书楠,席修璞,董 蕴,叶万海,张振东,郭 壮*
(1.湖北文理学院 化学工程与食品科学学院 鄂西北传统发酵食品研究所,湖北 襄阳 441053;2.襄阳市食品药品监督管理局,湖北 襄阳 441021)
作为中国地理标识产品和湖北省非物质文化遗产,湖北省枣阳市琚湾酸浆面以浆水、精制面条和香辣臊子等为主要成分,运用传统技艺加工制作而成。浆水是酸浆面品质形成的关键,一方面赋予了酸浆面独特的酸味,另一方面其浸泡发酵的芹菜亦是酸浆面中的佐料。浆水的制作环境较为开放,为了消除腐败菌的污染,通常会向前日剩余的浆水中加入等体积的沸水,并放置8~10 h后使用。经走访调研发现,酸浆面的制作和食用通常在每年的7~10月份,究其原因可能在于其他月份温度过低不利于微生物的发酵,从而无法制得浆水。在解析微生物群落结构的基础上,对浆水中优势菌群进行分离、鉴定、保藏和筛选,继而采用高密度发酵技术和冷冻干燥技术相结合的手段进行直投式发酵剂制备,同时辅以适宜的温度,可能对实现琚湾酸浆面的全天候乃至工业化生产具有积极意义。由此可见,对琚湾酸浆面浆水细菌多样性展开评价是极为必要的。
近年来兴起的Miseq高通量测序技术不仅具有通量高的优点[1],同时实现了多样本微生物群落结构的平行分析与评价[2],在酸奶[3]、腊肠[4]、葡萄酒[5]、窖泥[6]和泡菜[7]等发酵食品的微生物多样性研究中有着广泛的应用,为评价琚湾酸浆面浆水中细菌多样性提供了新的思路和技术手段。除此之外,在获取细菌16S rRNA序列信息的基础上,使用KOMODO(Known Media Database)等网站[8]对优势菌培养基的配方进行预测,对后续浆水中优势微生物菌株的分离亦可能具有积极的意义。
本项目以琚湾酸浆面浆水为研究对象,采用Miseq高通量测序技术对其细菌多样性进行了评价,同时结合生物信息学和多元统计学手段对微生物群落结构进行了平行分析。通过本研究的实施,以期全面解析酸浆面浆水中细菌的多样性,为后续琚湾酸浆面的全年化生产提供一定理论参考。
1 材料与方法
1.1 材料与试剂
乙二胺四乙酸二钠、异戊醇、氯仿、醋酸钠、三羟甲基氨基甲烷(Tris)饱和酚、乙醇、三羟甲基氨基甲烷、十六烷基三甲基溴化铵、十二烷基硫酸钠:国药集团化学试剂有限公司;QIAGEN DNeasy mericon Food Kit:德国QIAGEN公司;脱氧核糖核苷三磷酸(deoxy-ribonucleoside triphosphate,dNTP)、蛋白酶K、5×TransStartTM、FastPfu Buffer和FastPfu Fly DNA Polymerase:北京全式金生物技术有限公司;聚合酶链式反应(polymerase chain reaction,PCR)缓冲液:宝生物工程(大连)有限公司;338F/806R引物,其中正向引物中加入7个核苷酸标签(barcode):由武汉天一辉远生物科技有限公司合成。
1.2 仪器与设备
5810R台式高速冷冻离心机:德国Eppendorf公司;DYY-12电泳仪:北京六一仪器厂;Vetiri梯度基因扩增仪:美国AB公司;Miseq高通量测序平台:美国Illumina公司;R920机架式服务器:美国DELL公司;ND-2000C微量紫外分光光度计:美国Nano Drop公司;UVPCDS8000凝胶成像分析系统:美国BIO-RAD公司;2100芯片生物分析仪:美国Agilent公司。
1.3 试验方法
1.3.1 样品的采集
从湖北省枣阳市琚湾镇采集10个酸浆面浆水样品,浆水装入采样瓶中,置于含有冰袋的采样箱中低温运送回实验室,12 h内完成脱氧核糖核酸(deoxyribonucleic acid,DNA)提取工作。
1.3.2 DNA提取
50 mL浆水400 r/min离心10 min去除蔬菜残渣后,取上清液10 000 r/min继续离心10 min,使用QIAGEN DNeasy mericon Food Kit试剂盒对离心后得到的菌泥进行微生物总DNA提取。1.0%琼脂糖凝胶电泳检测条带明亮且无弥散现象,OD260nm/280nm在1.8~2.0且质量浓度>10 ng/μL的DNA样品置-20℃备用。
1.3.3 细菌16S rRNA聚合酶链式反应扩增及Miseq高通量测序
扩增体系为20 μL:10×PCR缓冲液4 μL,2.5 mmol/L dNTP2μL,5μmol/L正向和反向引物各0.8μL,5U/μLDNA聚合酶0.4 μL,DNA模板10 ng,其余部分为ddH2O。
扩增条件为:95℃变性3 min;95℃变性30 s,55℃退火30 s,72℃延伸45 s,30个循环;72℃延伸10 min。检测合格的PCR扩增产物稀释至100 nmol/L后,使用干冰寄往上海美吉生物医药科技有限公司进行Miseq高通量测序。
1.3.4 序列的拼接及质控
在序列进行拼接过程中同时进行质控,若成对序列重叠区碱基数<10 bp、最大错配比率>0.2、barcode存在碱基错配或引物碱基错配数>2 bp的序列均会予以剔除。将切除barcode和引物后的序列,合并为一个fna文件进行后续的生物信息学分析。
1.3.5 生物信息学分析
使用QIIME分析平台[9]结合已建立的脚本程序对1.3.4生成的fna文件进行序列的生物信息学分析:在使用Py-NAST[10]对序列校准排齐的基础上,进一步使用两步UCLUST法[11]归并建立分类操作单元(operational taxonomic units,OTU);从各OTU中选取代表性序列,使用RDP(Ribosomal Database Project,Release 11.5)[12]和Greengenes(Release 13.8)[13]数据库对序列进行同源性比对,通过对数据的整合进而明确其种属分类学地位;使用FastTree软件[14]构建系统发育进化树,进而对酸浆面浆水的Chao 1指数和Shannon指数等α多样性指标进行计算。
1.3.6 核酸登录号
本研究中所有序列数据已提交至MG-RAST数据库,ID号为mgp82912。
1.3.7 多元统计学分析
使用基于分类操作单元加权UniFrac距离的非加权组平均法(unweighted pair group method using arithmetic average,UPGMA)聚类对样品进行聚类分析,使用基于非加权UniFrac距离的主坐标分析法(principal coordinate analysis,PCoA)对样品进行空间排布,使用多元方差分析(multivariateanalysis of variance,MANOVA)对隶属于不同聚类样品间微生物群落结构的显著性进行分析,使用欧式距离对不同聚类样品间微生物群落结构的平均距离进行计算,使用冗余分析(redundancy analysis,RDA)对导致样品形成2个聚类的OTU进行甄别。使用Mega7.0(http://www.megasoftware.net/)进行系统发育树绘制,使用Origin 8.5软件和Matlab 2010b软件进行其他图的绘制。
2 结果与分析
2.1 序列丰富度和多样性分析
纳入本研究的10个酸浆面浆水样品16S rRNA测序情况及各分类地位数量如表1所示。
由表1可知,10个酸浆面浆水样品共产生了388 306条高质量16S rRNA序列,平均每个样品产生38 831条。本研究采用两步UCLUST归并法建立了分类操作单元,其中采用100%相似性聚类得到了129 865条代表性序列,在此基础上根据97%相似性聚类进一步得到了7 014个OTU,平均每个样品1 100个。由表1可知,样品X3具有最大的细菌微生物丰度,其Chao 1指数为4 282,而X6样品细菌微生物多样性最高,其Shannon指数高达6.63。
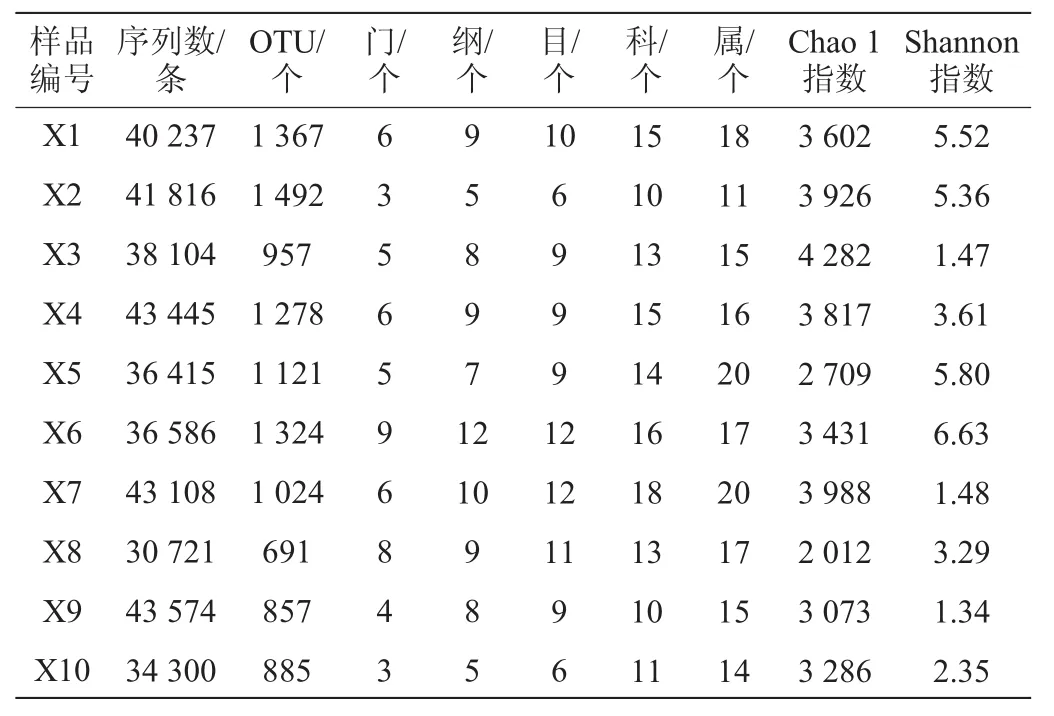
表1 样品16S rRNA测序情况及各分类地位数量Table 1 16S rRNA sequencing conditions and number of taxonomical levels
2.2 基于不同分类地位细菌菌群相对含量的分析
本研究产生的388306条序列,经同源性比对后,将其鉴定为11个门,20个纲,26个目,45个科和61个属,仅有0.036%的序列不能鉴定到属水平,其中有99.72%的细菌隶属于硬壁菌门(Firmicutes)的乳酸杆菌属(Lactobacillus)。由此可见,琚湾酸浆面浆水中的细菌几乎全部为乳酸杆菌。在得到7020个OTU的基础上,本研究进一步对平均相对含量>1.0%的OTU相对含量进行了分析,其结果如图1所示。
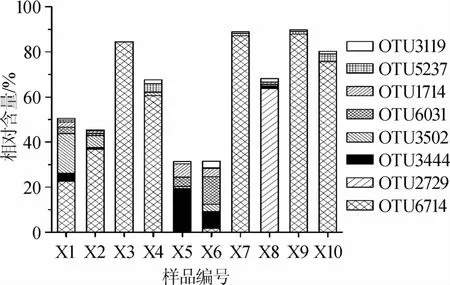
图1 酸浆面浆水样品中相对含量>1.0%的OTUFig.1 OTU with relative abundance more than 1.0%in Suanjiangmian Jiangshuisamples
由图1可知,酸浆面浆水中平均相对含量>1.0%的OTU包括OTU6714、OTU2729、OTU3444、OTU3502、OTU6031、OTU1714、OTU5237和OTU3119,8个OTU的累计平均相对含量高达63.79%。以Illumina MiSeq为代表的二代测序技术具有读数短的缺点,其无法完成16S rRNA的全长测序,因而为了保证结果的可靠性,一般仅在分类学地位“属”及以上水平对微生物的分类学地位进行确定。本研究进一步选取了8个平均相对含量>1.0%的OTU中的代表性序列,构建了系统发育树,其结果如图2所示。
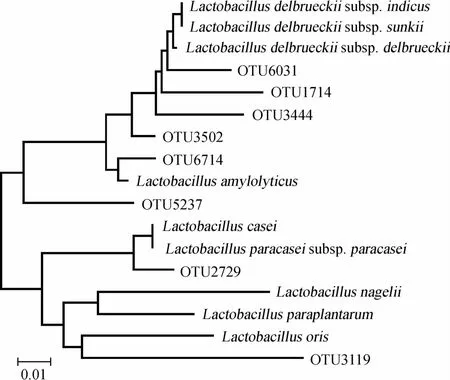
图2 相对含量>1.0%OTU的系统发育树Fig.2 Phylogenetic tree of OTU with relative abundance more than 1.0%
由图2可知,虽然不能将OTU鉴定到种水平,但OTU6031、OTU1714、OTU3444和OTU3502可以与德氏乳杆菌(Lactobacillusdelbrueckii)形成一个聚类,OTU6714与嗜淀粉乳杆菌(L.amylolyticus)形成一个聚类,OTU2729可以与干酪乳杆菌(L.casei)和副干酪乳杆菌(L.paracasei)形成一个聚类,OTU3119可以与口乳杆菌(L.oris)形成一个聚类。如果一个OTU在10个酸浆面浆水样品中均存在,则将其定义为核心OTU。本研究进一步统计了OTU在10个酸浆面浆水样品中出现的次数,其结果如图3所示。
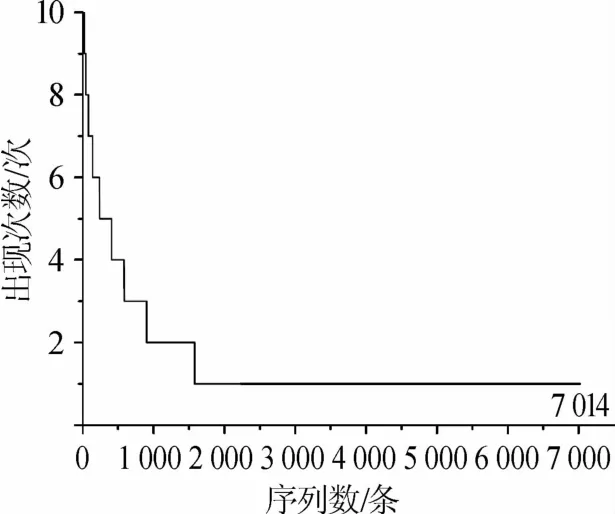
图3 OTU在10个样品中出现次数统计Fig.3 Occurrence frequency statistics of OTU in 10 samples
由图3可知,10个样品共有18个核心OTU,虽然仅占OTU总数的0.26%,但其包含了212 778条序列,占所有质控后合格序列数的54.80%。此外,在10个样品中出现9次、8次、7次和6次的OTU各有23个、39个、57个和99个,分别占OTU总数的0.33%、0.56%、0.81%和1.41%,4类OTU共包含95 555条序列,占所有质控后合格序列数的24.61%。虽然在10个样品中仅出现1次的OTU多达5 435个,占到OTU总数的77.49%,但其仅包含12 967条序列,平均每个OTU包含2.4条序列。由此可见,在OTU水平上,10个酸浆面浆水样品共有大量的细菌类群,其累计平均相对含量达到54.80%。本研究进一步对18个核心OTU进行了分析,其在各酸浆面浆水样品中相对含量的热图如图4所示。
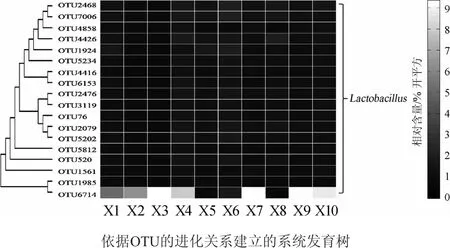
图4 核心OTU在各样品中相对含量的热图Fig.4 Heat map of the relative contents of core OTUs in 10 samples
由图4可知,18个核心OTU均隶属于乳酸杆菌属(Lactobacillus),除OTU6714的平均相对含量达45.70%外,其他核心OTU的平均相对含量均<1.0%。值得一提的是,不同样品中OTU6714的含量亦存在较大的差异,其在X3、X7和X9样品中达87%,而在X5和X8样品中的含量均<0.01%。2.3酸浆面浆水细菌菌群群落结构的研究
在对酸浆面浆水中不同分类地位细菌菌群相对含量进行分析的基础上,本研究进一步采用UPGMA聚类分析和主坐标分析对其细菌菌群群落结构进行了研究,基于分类操作单元加权UniFrac距离的UPGMA聚类分析如图5所示。
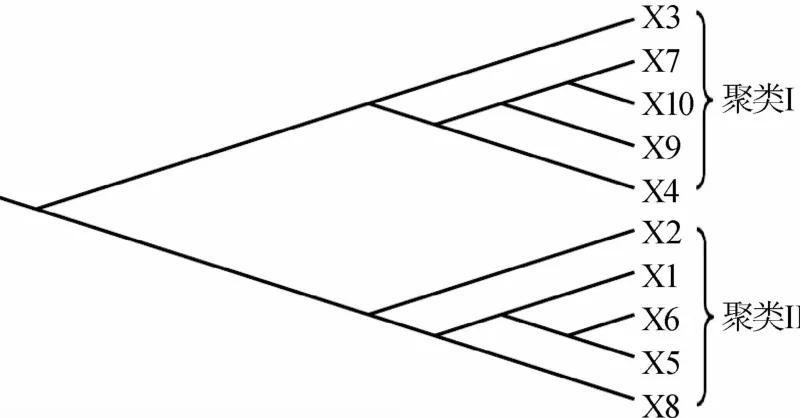
图5 基于分类操作单元加权UniFrac距离的UPGMA聚类分析Fig.5 UPGMA clustering analysis of OTUs based on weighting UniFrac distance
由图5可知,10个样品整体上可以分为2个聚类,其中聚类I由样品X3、X4、X7、X9和X10构成,聚类II由样品X1、X2、X5、X6和X8构成。由此可见,虽然若干酸浆水样品存在大量的核心细菌菌群,但某些样品间微生物的群落结构存在较大的差异。本研究进一步使用基于非加权UniFrac距离的主坐标分析对10个样品的空间排布进行了研究,结果如图6所示。
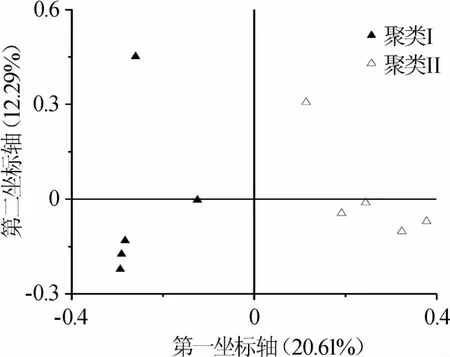
图6 基于分类操作单元加权UniFrac距离的主坐标分析Fig.6 Principal coordinate analysis of OTUs based on weighting UniFrac distance
由图6可知,隶属于不同聚类的酸浆面浆水样品在空间排布上呈现出明显的分离趋势,这进一步证实了隶属于聚类I和聚类II的样品其微生物群落结构构成存在较大的差异,通过MANOVA验证发现两者差异极显著(P<0.001)。值得一提的是,本研究进一步采用欧式距离对两个聚类样品间微生物群落结构的平均距离进行了计算,结果发现隶属于聚类I的样品组间平均距离为0.825±0.020,而聚类II为0.815±0.025,经显著性分析发现两者差异不显著(P>0.05)。
虽然隶属于不同聚类的酸浆面浆水其微生物群落结构明显不同,然而究竟是由于那些OTU不同导致了该现象的发生是本研究亟待解决的问题。通过设置聚类I/聚类II作为起约束作用的解释变量,本研究进一步使用RDA用于预测和解释全部平均含量>1.0%的OTU数据组成的响应变量,从而对能显著解释不同聚类间酸浆面浆水细菌微生物群落结构存在差异的最小变量组合进行确定[15]。经分析发现,数据中有19.3%的变异度能够被聚类I/聚类II分组所解释,而通过蒙特卡罗置换检验发现这一约束因素具有显著性(P=0.024)。RDA双序图如图7所示。
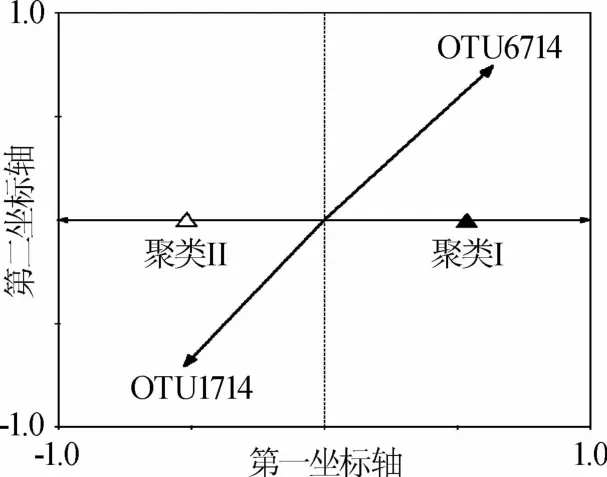
图7 冗余分析双序图Fig.7 Biplot of the redundancy analysis
由图7可知,在平均相对含量>1.0%的OTU中,OTU6714和OTU1714与RDA排序图约束轴上的样本赋值良好相关,2个OTU均隶属于乳酸杆菌属(Lactobacillus)。由此可见,正是由于上述2个OTU在不同样品中的分布及含量不同,进而导致了隶属于聚类I和聚类II的样品其微生物群落结构明显不同。由图7可知,在RDA排序图中OTU6714位于图的右侧(即聚类I)而OTU1714位于图的左侧(即聚类II),这说明OTU6714的相对含量在隶属于聚类I的样品中较高,而OTU1714呈现出相反的趋势,通过Mann-Whitney验证发现2个OTU在不同聚类的酸浆面浆水样品间差异非常显著(P<0.01)。
3 结论
在进行微生物基因组DNA提取的基础上,本研究使用高通量测序技术,对琚湾酸浆面这一中国地理标识产品的浆水细菌多样性进行了评价。结果发现,琚湾酸浆面浆水中的细菌微生物中乳酸杆菌相对含量达99.72%,虽然不同样品共有54.80%的核心细菌类群,但由于部分乳酸杆菌属细菌的不同导致其可以划分为两个聚类,且隶属于不同聚类的样品细菌群落结构存在显著的差异。通过本研究的开展,可为后续酸浆面浆水用发酵剂的制备提供一定理论参考,进而使酸浆面的全年化生产成为可能。
[1]CAPORASO J G,LAUBER C L,WALTERS W A,et al.Ultra-highthroughput microbial community analysis on the Illumina HiSeq and MiSeq platforms[J].ISME J,2012,6(8):1621-1624.
[2]ROH S W,KIM K H,NAM Y D,et al.Investigation of archaeal and bacterial diversity in fermented seafood using barcoded pyrosequencing[J].ISME J,2010,4(1):1-16.
[3]智楠楠.Illumina Miseq平台深度测定酸奶中微生物多样性[J].食品工业科技,2016,37(24):78-82.
[4]JUSTYNA P,ANNALISA R,VINCENZA P,et al.Bacterial diversity in typical Italian salami at different ripening stages as revealed by highthroughput of 16s rDNA amplicons[J].Food Microbiol,2015,46(4):342-356.
[5]BOYNTON P J,GREIG D.Fungal diversity and ecosystem function data from wine fermentation vats and microcosms[J].Data Brief,2016,8(12):225-229.
[6]HU X,DU H,REN C,et al.Illuminating anaerobic microbial community and cooccurrence patterns across a quality gradient in Chinese liquor fermentation pit muds[J].Appl Environ Microbiol,2016,82(8):2506-2515.
[7]YANG H,WU H,GAO L,et al.Effects ofLactobacillus curvatusand Leuconostoc mesenteroideson Suan Cai fermentation in northeast China[J].J Microbiol Biot,2016,26(12):2148-2158.
[8]OBERHARDT M A,ZARECKI R,GRONOW S,et al.Harnessing the landscape of microbial culture media to predict new organism-media pairings[J].Nat Commun,2015,6:8493.
[9]CAPORASO J G,KUCZYNSKI J,STOMBAUGH J,et al.QIIME allows analysis of high-throughput community sequencing data[J].Nat Meth,2010,7(5):335-336.
[10]CAPORASO J G,BITTINGER K,BUSHMAN F D,et al.PyNAST:a flexible tool for aligning sequences to a template alignment[J].Bioinformatics,2010,26(2):266-267.
[11]EDGAR R C.Search and clustering orders of magnitude faster than BLAST[J].Bioinformatics,2010,26(19):2460-2461.
[12]COLE J R,CHAI B,FARRIS R J,et al.The ribosomal database project(RDP-II):introducing myRDP space and quality controlled public data[J].Nucleic Acids Res,2007,35(1):169-172.
[13]DESANTIS T Z,HUGENHOLTZ P,LARSEN N,et al.Greengenes,a chimera-checked 16S rRNA gene database and workbench compatible with ARB[J].Appl Environ Microbiol,2006,72(7):5069-5072.
[14]PRICE M N,DEHAL P S,ARKIN A P.Fasttree:computing large minimum evolution trees with profiles instead of a distance matrix[J].Mol Biol Evol,2009,26(7):1641-1650.
[15]WANG T,CAI G,QIU Y,et al.Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers[J].ISME J,2012,6(2):320-329.
