气相色谱-串联质谱法测定山药中农药残留的一测多评研究
2018-01-30余敏灵
陈 岚 ,余敏灵 ,杨 佳 ,梁 华 ,李 根
(1.四川省成都市妇女儿童中心医院,四川 成都 610091; 2.四川省乐山市食品药品检验检测中心,四川 乐山 614000)
近年来,随着中药材的产业化发展,农药污染问题日益严重,农药残留问题始终制约着中药走向国际化。2015年版《中国药典(四部)》收载了多种有机氯类、有机磷类等农药残留的气相色谱检查方法[1]。文献中农药残留检测方法多以气相色谱(GC)法、液相色谱法、气相色谱-质谱(MS)联用及液相色谱-质谱联用为主[2-8],但都存在对照品获得困难、操作烦琐或多指标检测成本高的问题。为此,本研究中应用气相色谱-串联质谱(GC-MS/MS)法,根据色谱保留时间和质荷比信息对样品进行定性分析,以联苯菊酯二级离子扫描色谱峰面积为参照,计算其与甲氰菊酯二级离子扫描色谱峰面积和溴氰菊酯二级离子扫描色谱峰面积的校正因子,建立GC-MS/MS法测定山药[1]中联苯菊酯等3种农药残留的一测多评方法,结果满意,可为GS-MS/MS测定农药残留量提供一测多评[9-12]研究思路。现报道如下。
1 仪器与试药
1.1 仪器
GCMS-TQ8030型气质联用仪,Rxi-5SilMS型毛细管柱(30 m × 0.25 mm,0.25 μm),工作站为 LabSolutions GCMS Solutions(4.20 版,岛津中国科技有限公司);Cleanert TPH净化柱(天津博纳艾洁尔公司)。
1.2 试药
联苯菊酯、甲氰菊酯、溴氰菊酯标准溶液(规格为100 μg/mL,批号分别为 GSB05-2333-2016,GSB05-2306-2016,GSB05-2310-2016);乙腈、丙酮为色谱纯,氯化钠、无水硫酸钠均为优级纯。山药样品采购于乐山中药材有限公司,按2015年版《中国药典(一部)》所载方法鉴定为薯蓣 Dioscorea opposita Thunb.的干燥根茎[13],样品批号分别为 20161001,20161002,20161003。
2 方法与结果
2.1 测定条件
2.1.1 气相色谱条件
色谱柱:采用 Rxi-5SilMS型毛细管柱(30 m×0.25 mm,0.25 μm);进样口温度:250 ℃ ;线速度:47.2 cm /s;程序升温:50 ℃保持 1 min,以 25 ℃ /min 的速率升至125℃,再以10℃ /min的速率升至300℃,保持15 min;进样体积:1 μL;载气:高纯度氦气(>99.999%)。
2.1.2 质谱条件
离子源温度:200℃;接口温度:250℃;溶剂延迟时间:5 min;碰撞气:氩气(纯度大于 99.999% );电离方式:见表 1。

表1 3种农药的质谱电离方式
2.2 溶液制备
标准贮备溶液:精密量取联苯菊酯、甲氰菊酯、溴氰菊酯标准溶液各1 mL,置10 mL容量瓶中,加丙酮稀释至刻度,摇匀,作为标准贮备溶液(每1 mL含联苯菊酯、甲氰菊酯、溴氰菊酯各 10 μg)。
样品溶液:取山药(批号为20161001)约50 g,粉碎成粉末(过 3号筛),称取粉末 1.023 1 g,精密称定,置50mL离心管中,加入15 mL乙腈,以15000 r/min的速率匀浆提取1 min,加入2 g氯化钠,再匀浆提取1 min,以4 200 r/min的速率离心5 min,取上清液置150 mL鸡心瓶中,再向离心管中加入15 mL乙腈,按上述方法重复匀浆提取1次,合并提取液,于40℃水浴旋转蒸发至约1 mL。Cleanert TPH固相萃取柱上加入约2 cm高无水硫酸钠,用10 mL正己烷 -丙酮(6∶4)预洗后,将样品浓缩液移入柱中,用 25 mL正己烷 -丙酮(6∶4)洗脱,收集洗脱液于40℃水浴旋转浓缩至近干,用丙酮定容至 1 mL,0.2 μm 滤膜滤过,即得。
空白溶液:除称取样品粉末外,按上述操作制备空白溶液。
样品加标溶液:称取同一批(批号为20161001)山药粉末1.031 0 g,精密称定,按样品溶液的制备方法,自“置50 mL离心管中”起到“收集洗脱液于40℃水浴旋转浓缩至近干”,用丙酮转移至1 mL容量瓶中,另取标准贮备溶液1 mL置10 mL容量瓶中,用丙酮稀释至刻度,摇匀,精密量取此溶液5 μL置上述1 mL容量瓶中,用丙酮定容至刻度,摇匀,用0.2 μm滤膜过滤,即得。
2.3 图谱解析
精密量取标准贮备溶液 25 μL,置 5 mL容量瓶中,加丙酮稀释至刻度,摇匀,作为对照品溶液。按拟订测定条件,取对照品溶液、样品溶液、样品加标溶液、空白溶液分别进样,记录色谱图。结果见图1和图2。在对照品溶液二级离子扫描色谱图中有联苯菊酯(181.1 /166.1,即母离子与子离子的 m /z值之比,下同)、甲氰菊酯(265.1 /210.1)和溴氰菊酯(252.9 /93.0)色谱峰;保留时间,联苯菊酯为17.514 min,甲氰菊酯为17.695 min,溴氰菊酯为 21.783 min;在空白溶液色谱图和样品溶液色谱图中无上述色谱峰,在样品加标溶液色谱图中有和对照品溶液色谱图中保留时间一致的联苯菊酯、甲氰菊酯和溴氰菊酯二级离子扫描色谱峰。对照品溶液和样品加标溶液的二级离子碎片图谱中,联苯菊酯在 153.1,166.1,179.1 处(丰度比为 5 ∶100 ∶19,以166.1 为参比),甲氰菊酯在 89.0,172.1,210.1 处(丰度比为 40 ∶14 ∶100,以 210.1 为参比),溴氰菊酯在 77.0,93.0,171.9 处(丰度比为 22 ∶100 ∶84,以 93.0 为参比)均有二级离子碎片峰。

图1 二级离子扫描色谱图
2.4 方法学考察
线性关系考察:精密量取标准贮备溶液5,10,20,25,30,40,50 μL,分置 7 个 5 mL 容量瓶中,加丙酮稀释至刻度,摇匀。按拟订测定条件进样测定,以组分二级离子扫描(181.1 /166.1)峰面积(A)对进样量(m,ng)进行线性回归,得回归方程。结果见表2。
精密度试验:精密量取标准贮备溶液 20 μL,置5 mL容量瓶中,加丙酮稀释至刻度,摇匀。按拟订测定条件重复进样6次。测定结果见表2。结果表明,仪器精密度好。
重复性试验:精密量取标准贮备溶液20 μL,分别置6个5 mL容量瓶,加丙酮稀释至刻度,摇匀。按拟订测定条件分别进样测定。结果见表2。可见,方法重复性好。
样品溶液稳定性试验:取样品加标溶液,按拟订测定条件,分别在 0,2,4,8,12,14,16,20,24 h 时进样。结果联苯菊酯、甲氰菊酯、溴氰菊酯二级离子扫描色谱峰面积变化不大,RSD 分别为 1.12% ,1.25% ,0.93%(n=9),表明样品溶液在24 h内稳定。
检出限与定量限确定:取样品加标溶液,按拟订测定条件进行测定,测得信噪比(S/N),以 S/N=3计算最低检出限,S/N=10计算最低定量限。结果见表2。
回收率试验:取2.2项下山药粉末9份,分为3组,每组3份,每份约1 g,精密称定,分置9个50 mL离心管中,每组分别加入标准贮备溶液 4,5,6 μL,依法制备样品溶液,取样品溶液,按拟订测定条件分别进样,记录图谱。按回归方程 A=10 388 461 m-236计算供试品中联苯菊酯含量,按校正方程 A甲=10388461 mf甲/联-236计算甲氰菊酯含量,按校正方程 A溴=10 388 461 mf溴/联-236计算溴氰菊酯含量。结果见表3。

图2 二级离子碎片图谱

表2 线性关系考察、精密度、重复性及检出限与定量限试验结果
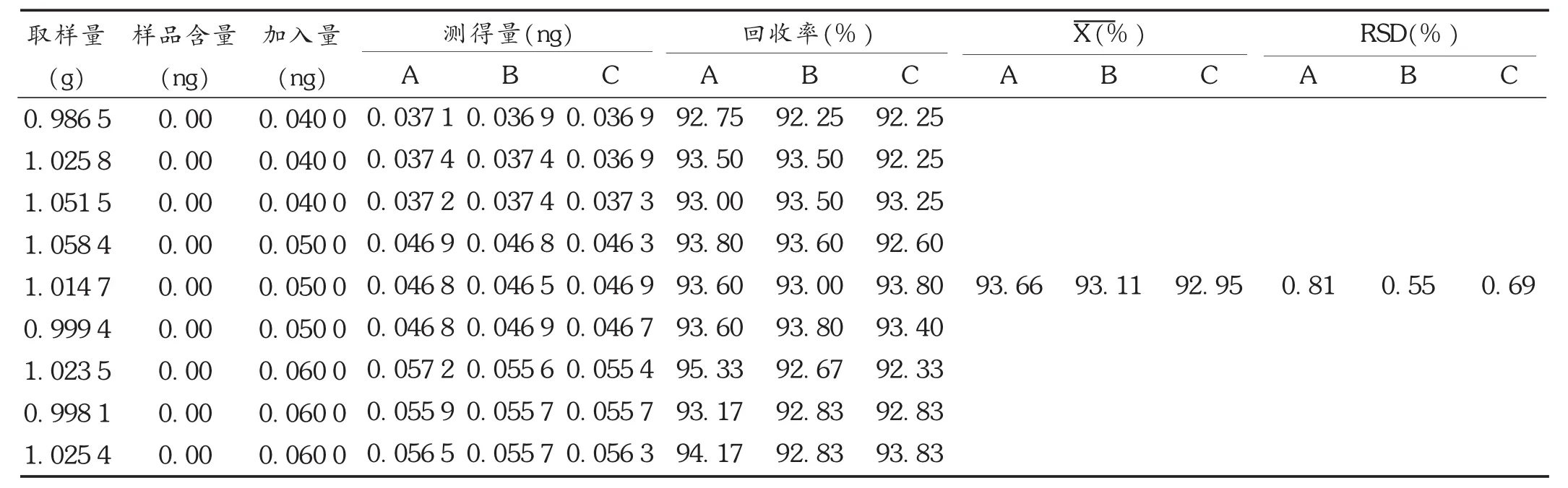
表3 加样回收试验结果(n=9)
f值和 t值测定:甲氰菊酯与联苯菊酯色谱峰峰面积的校正因子 f甲/联=523 142/10 388 461=0.050 4,相对保留时间 t甲/联=17.695 /17.514 =1.010 3;溴氰菊酯与联苯菊酯色谱峰峰面积的校正因子 f溴/联=520 881/10 388 461=0.050 1,相对保留时间 t溴/联=21.783 /17.514 = 1.243 7。
f值耐受性考察:实际应用中,色谱条件可能会有一些变化,故对线速度、进样体积等可能影响 f值的因素进行考察,结果 f值的波动不大。详见表4。
2.5 样品中农药残留量测定
分别取3批山药样品约50 g,分别粉碎成粉末(过3号筛),分别取粉末约1 g,精密称定,依法制备样品溶液,取样品溶液按拟订测定条件分别进样,记录图谱,计算样品中联苯菊酯、甲氰菊酯、溴氰菊酯残留量。结果见表5。
3 讨论
以联苯菊酯为对照品测定样品中3种农药残留,其原理是利用样品色谱图中联苯菊酯、甲氰菊酯和溴氰菊酯的色谱峰相对保留时间进行定位[2],以及二级离子碎片图谱碎片离子峰和丰度比对联苯菊酯、甲氰菊酯、溴氰菊酯色谱峰进行定性分析。定量分析,通过精密量取联苯菊酯、甲氰菊酯和溴氰菊酯对照品溶液各适量,配制成不同浓度的混合溶液,按设定条件进样,记录色谱图,绘制进样量对其峰面积的回归曲线,以联苯菊酯回归曲线斜率与甲氰菊酯、溴氰菊酯回归曲线斜率的比值计算校正因子(fsk=Ks/Kk),通过联苯菊酯回归曲线计算联苯菊酯含量,通过校正因子及联苯菊酯回归曲线计算甲氰菊酯、溴氰菊酯含量。

表4 相对校正因子耐受性考察结果

表5 3批样品中3种农药残留量测定结果(ng/kg)
GC-MS联用法和GC-MS/MS法既具有GC的高分离效能,又具有MS可鉴定化合物结构的特点,可同时快速测定样品中多种残留农药及其衍生物。本研究中采用的一测多评法,通过设定1种对照品,建立该成分与其他成分间的相对响应因子,再用该因子计算其他成分含量。一测多评法具有成本低、效率高等优点,能有效解决多指标质量控制面临的对照品短缺、不易获得、检测成本相对昂贵和农药对照品造成环境污染等问题,是较常用的一种质量控制方法。然而,中药材往往含有多种成分,且其中某些成分可能对测定产生干扰。因此,使用一测多评法对中药材残留农药进行含量测定,尤其在测定某一具体品种时应设计具体的试验参数,并进行方法学验证。
校正因子计算法常应用于化学药中有关物质的测定,以及中药材及其复方制剂中多指标成分的测定。校正因子的表示方法很多,本试验中是指气相色谱法的相对质量校正因子。测定时,可精密称(量)取1个成分对照品和其他物质对照品各适量,分别配制成不同浓度的溶液,进样,记录色谱图,绘制选定成分进样量和其他成分进样量对其峰面积的回归曲线,以其回归直线斜率之间的比值计算。
本试验中采用乙腈提取,经Cleanert TPH专用净化柱净化,根据色谱相对保留时间和质荷比信息对样品进行定性分析,以联苯菊酯二级离子扫描峰面积为参照,成功建立了以联苯菊酯为对照品测定山药中联苯菊酯等3种农药残留量的GC-MS/MS方法。一般而言,当色谱条件有细微改变时,相对保留时间有细微变化,故需考察该时间耐受性。本试验中未作考察,是因为被测定物质的定性主要采用二级离子碎片峰及其丰度比确认;又因乙腈对农药溶解度较大,可溶入的杂质量适中,故选择乙腈为提取液。另外,本试验中还在Cleanert TPH固相萃取柱上加入约2 cm高的无水硫酸钠,以除去样品溶液及洗脱液中的水分,从而充分净化样品,进一步保证检测质量。
综上所述,本研究中建立的一测多评法简单,快速,定量准确,适用于山药中联苯菊酯、甲氰菊酯、溴氰菊酯农药残留的测定。
[1]国家药典委员会.中华人民共和国药典(四部)[M].北京:中国医药科技出版社,2015:209 -223.
[2]张 羽中,郎 朗.中药材中农药残留分析的研究进展[J].黑龙江医药,2015,28(2):235 - 238.
[3]孔令军,张娅婷,谷令彪,等.中药材农药残留的研究进展[J].中国实验方剂学杂志,2015,21(21):231 - 234.
[4]傅巧真,路俊仙,王 萌,等.中药材农药残留原因及防治措施的研究进展[J].时珍国医国药,2014,25(4):925 -927.
[5]刘 芳,欧阳慧子,柴士伟,等.GC法检测72种中药饮片有机氯农药残留量[J].中国药房,2016,27(36):5147 - 5150.
[6]殷玉洁,夏秀萍,毛福英,等.药用植物中农药残留检测技术的研究进展[J].中国药房,2017,28(12):1721 - 1726.
[7]李冬华,马 潇,赵建邦,等.我国市售药材中64种农药残留的本底调查与检测分析[J].西部中医药,2011,24(12):7-10.
[8]刘 姣,刘雪峰,樊宝娟.GC-MS法测定18种药材中的农药残留量[J].西北药学杂志,2017,32(1):9 -13.
[9]徐赛华,沈昱翔.一测多评法测定重楼药材中4种皂苷类成分含量[J].医药导报,2017,36(9):1029 - 1033.
[10]王 欣,覃 瑶,孙建彬,等.一测多评法测定黄连须中的5种生物碱[J].华西药学杂志,2017,32(4):417 -420.
[11]于雪娥,秦建平,李家春,等.一测多评法同时测定淫羊藿总黄酮胶囊中7种黄酮类成分[J].中国实验方剂学杂志,2017,23(7):79 - 85.
[12]刘 峰,马久太,王浩仁,等.一测多评法测定沙棘鲜果中槲皮素、山柰素和异鼠李素含量[J].中国药业,2017,26(13):24 - 27.
[13]国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2015:28.
