噁唑烷酮—喹诺酮杂合物的研究进展
2018-01-26刘利利张继瑜
刘利利,张继瑜
(中国农业科学院兰州畜牧与兽药研究所,农业部兽药创制重点实验室,兰州 730050)
尽管抗生素的发现为医疗革命开启了新的篇章,然而随着抗生素在全世界范围的普及和不断被滥用,无论是革兰阳性菌还是革兰阴性菌均已出现了严重的耐药性,同时还出现了多重耐药的“超级细菌”,抗生素似乎逐渐失去了往日的辉煌。据世界卫生组织(WHO)统计,每天约有5万患者死于感染性疾病,感染性疾病已经成为人类健康与社会发展的严重威胁,因此引起了人们的高度重视。全球范围内出现的耐甲氧西林的金黄色葡萄球菌(methicillin resistantStaphylococcusaureus, MRSA)[1]和表皮葡萄球菌(methicillin resistantStaphylococcusepidermidis, MRSE)[2]、耐青霉素的肺炎链球菌(penicillin resistantStreptococcuspneumoniae, PRSP)[3]以及耐万古霉素肠球菌(vancomycin resistantEnterococcus, VRE)[4]是当前临床中存在的主要耐药菌株,尤其是耐万古霉素肠球菌(VRE)的出现,突破了严重感染患者治疗的“最后手段”,已经成为引起医院内感染的主要原因。
面对日益严重的耐药性问题,世界上许多制药公司和研究机构都在积极寻找能够突破这种困境的新型高效的抗感染药物。随着有机化学、生物化学以及分子药理学的快速发展,利用“拼合原理”将两种药物的结构或者药效基团嵌合到一起所形成的杂交分子能够扩大药物的抗菌谱、增强抗菌活性,对耐药株敏感同时能降低细菌耐药的概率[5],具有很大的优势,已经成为近些年来研发新型抗菌药物的热点。
1 噁唑烷酮类抗菌药
早在20世纪80年代,杜邦(DuPont)公司发现了两个具有潜在应用价值的噁唑烷酮类化合物DUP721和DUP105[6],但因其毒性较大而终止了后续研究。20世纪90年代,Pharmacia &Upjohn 公司在此基础上经过化学结构改造,得到了两个新化合物:依哌唑胺(eperezolid)和利奈唑胺(linezolid,结构式见图1),其中利奈唑胺已通过了FDA批准,于2000年4月首次在美国上市,成为了第一个面向市场的噁唑烷酮类抗菌药[7-8]。泰地唑胺(tedizolid,结构式见图2)为第二代噁唑烷酮类抗生素,已于2014年6月20日通过了美国FDA的批准,得以上市[9-10]。此外,目前还有3个噁唑烷酮类化合物进入了Ⅰ期临床研究,分别是Astrazeneca公司开发的AZD2563[11]、印度公司开发的RBx-7644(Ranbezolid hydroch-loride)[12]、Pharmacia公司开发的VRC-3783[13]。
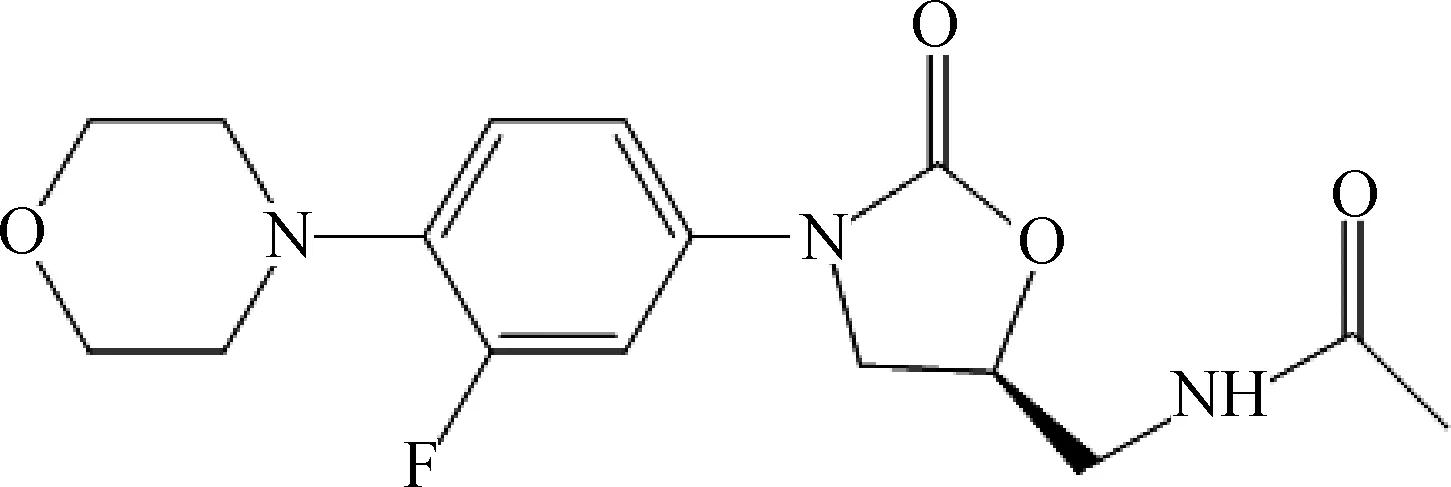
图1 利奈唑胺的结构式Fig.1 The chemical structure of linezolid
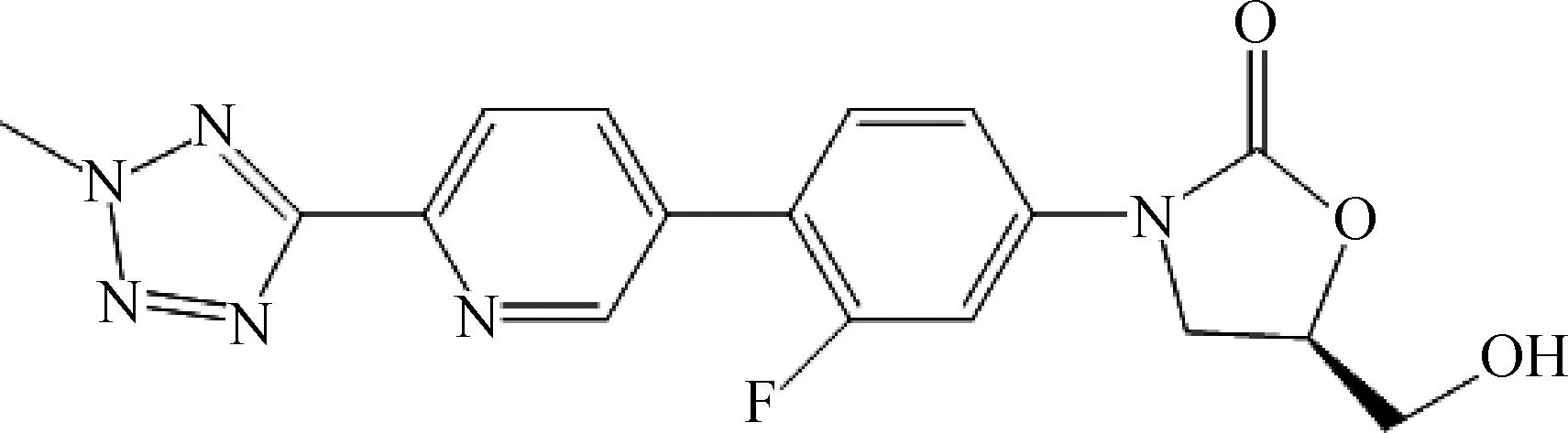
图2 泰地唑胺的结构式Fig.2 The chemical structure of tedizolid
噁唑烷酮类抗菌剂是一类化学全合成的新型抗菌剂,通过抑制细菌蛋白质的合成而达到抗菌效果,对G+菌及其耐药菌的感染有很好的疗效[14]。药物化学家对该类化合物的构效关系进行了总结(图3),大致如下:(1)环A、B之间不应有基团;(2)噁唑烷酮母核5位与乙酰胺基之间最适碳链长度为一个碳原子;(3)噁唑烷酮母核5位碳原子应为S构型;(4)噁唑烷酮母核2位与乙酰胺基上的羰基,噁唑烷酮母核1位氧原子以及酰胺N上的H为活性必需基团;(5)苯环3′或3′,5′氟取代活性最好[15-17]。
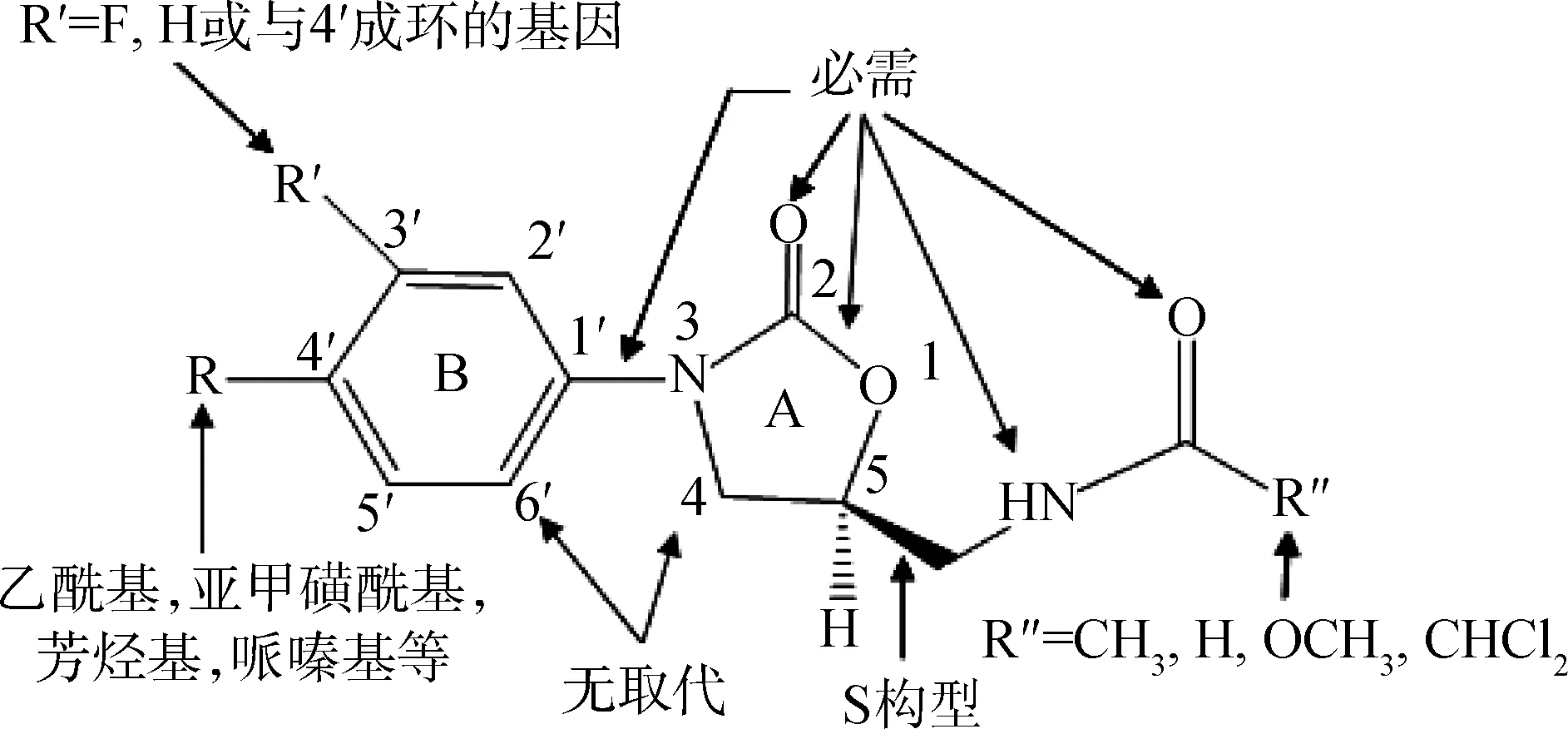
图3 噁唑烷酮类抗菌药的构效关系Fig.3 Structure activity relationships of the oxazolidinone antibiotics
2 喹诺酮类抗菌药
自1962年美国Sterling-Winthrop研究所G. Y. Lesher等发现第一个喹诺酮类抗菌药——萘啶酸以来[18],在短短的50年里,喹诺酮类已经发展为一大类抗菌谱广、抗菌活性强的抗菌药。因其良好的抗感染作用,已经成为头孢类抗生素之后全球抗感染市场用药量最大的药物。现在喹诺酮类药物已发展至第四代,分别以萘啶酸、吡哌酸、氧氟沙星、克林沙星等为代表,主要作用于G-菌。其中,第三代喹诺酮类药物——氟喹诺酮类药物,是此类药物发展的高峰期,也是目前市场上应用最多的一类药物。该类药物主要通过作用于细菌DNA回旋酶和拓扑异构酶Ⅳ,阻碍细菌DNA复制而发挥抑菌作用[19]。喹诺酮类药物(母核结构见图4)构效关系如下:(1)环A为抗菌必需基团,环B可以是苯环、吡啶环、嘧啶环等;(2)3位羧基和4位羰基为活性必需基团;(3)1位取代以乙基、氟乙基和环丙基为最好;(4)5位以氨基取代为最好;(5)6位取代活性顺序为F>Cl>CN≥NH2≥H;(6)7位取代活性顺序为哌嗪基≥甲氨基>卤素>氢;(7)8位引入氟活性最佳,但光毒性也增加;(8)1位和8位成环时,产生光学结构以S型为好[20-21]。

图4 喹诺酮类药物母核结构Fig.4 The chemical structure of quinolones’ mother nucleus
3 噁唑烷酮—喹诺酮杂合物
抗生素耐药性在我国尤为严重,目前几乎所有常见的感染致病菌均出现了耐药性,尤其是革兰阳性菌[22],因此急需寻找一种新的策略来应对当前的困境。除了寻找作用于新靶点的抗菌药外,将两类抗生素的药效基团拼合到一个分子当中的策略似乎是一个不错的选择。喹诺酮类药物具有双靶点作用机制且抗菌活性强,同时化学稳定性好,在化学合成方面易操作,因此成为拼合抗菌药物研发的首选对象。目前国外已经对许多喹诺酮类拼合抗菌化合物进行了研究,如与甲氧苄啶、利福霉素类、氨基糖苷类等形成的拼合抗菌药物,具有广阔的应用开发前景[23-25]。噁唑烷酮类抗菌药是一类很有前景的全新结构抗菌剂,与喹诺酮类药物作用机制和抗菌谱均不同,因此将噁唑烷酮类与喹诺酮类的药效基团进行拼合形成的杂合化合物所具有的双作用机制不仅可以扩大抗菌谱,还可能克服细菌耐药性问题,有很大的发展空间。
3.1 噁唑烷酮—喹诺酮杂合物的设计及合成
噁唑烷酮类和喹诺酮类构效关系表明在噁唑烷酮类苯基4位和喹诺酮类苯基7位进行结构改造,对其抗菌活性影响不大,而且在噁唑烷酮类苯基的4位和喹诺酮类苯基的7位都含有哌嗪基或者氮杂环,因此两类抗菌剂的拼合多是将噁唑烷酮类苯基4位和喹诺酮类苯基7位通过间隔基(X)连接在一起,形成噁唑烷酮—喹诺酮杂合物Ⅰ(图5)[26-29]。克里斯蒂安·哈伯舒兰等还采用另一种拼合方式经间隔基合成了噁唑烷酮—喹诺酮杂合物Ⅱ(图6)[30]。此外,也有将喹诺酮的1位N与噁唑烷酮苯基的7位进行直接拼合的实例,但抗菌活性消失[27]。
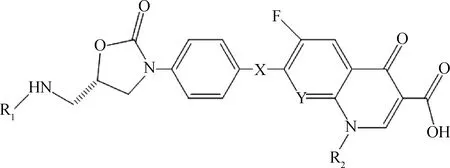
图5 噁唑烷酮—喹诺酮杂合物Ⅰ的结构式Fig.5 The chemical structure of oxazolidinone-quinolone hybrids Ⅰ

图6 噁唑烷酮—喹诺酮杂合物Ⅱ的结构式Fig.6 The chemical structure of oxazolidinone-quinolone hybrids Ⅱ
3.2 噁唑烷酮—喹诺酮杂合物的构效关系
在噁唑烷酮—喹诺酮杂合物Ⅰ中,间隔基X是影响杂合物活性的重要部位,通过修饰间隔基X可获得一系列噁唑烷酮—喹诺酮杂合物,其抗菌活性的测试结果见表1。从表1可以看出,除杂合物i和q外,其余杂合物均有很好的抗菌作用[26-28]。当间隔基为哌嗪基时(b),抗菌谱与利奈唑胺相似,有很强的噁唑烷酮类活性,同时保留了部分喹诺酮活性,对环丙沙星耐药株有效,但对利奈唑胺耐药株作用较弱[26-28];当间隔基为3-氨基-氮杂环丁烷基或氨基吡咯烷基时(d、e),抗菌活性与喹诺酮更相似,对G-菌和利奈唑胺耐药株更敏感,但对环丙沙星耐药株的作用较弱[26,28];当间隔基为3/4-羟基-含氮杂环/吡咯烷基(f、g、h、j),双作用机制较为平衡,对环丙沙星耐药株和利奈唑胺耐药株均敏感[28];吡咯烷基3位的立体化学对活性也有影响,杂合物f和g相比可知3位为S构型时(g)喹诺酮活性更占优势,而杂合物n和o相比表明3位为R构型时(o)喹诺酮活性稍微增强,为S构型时(n)噁唑烷酮活性稍微增强[28];间隔基长度对抗菌活性的影响并不显著(h/k/l、m/p)[28];低pH环境能在一定程度上增强杂合物的抗菌活性[26];喹诺酮部分3位羧基成脂,抗菌活性几乎消失[27]。
表1噁唑烷酮—喹诺酮杂合物的抗菌活性
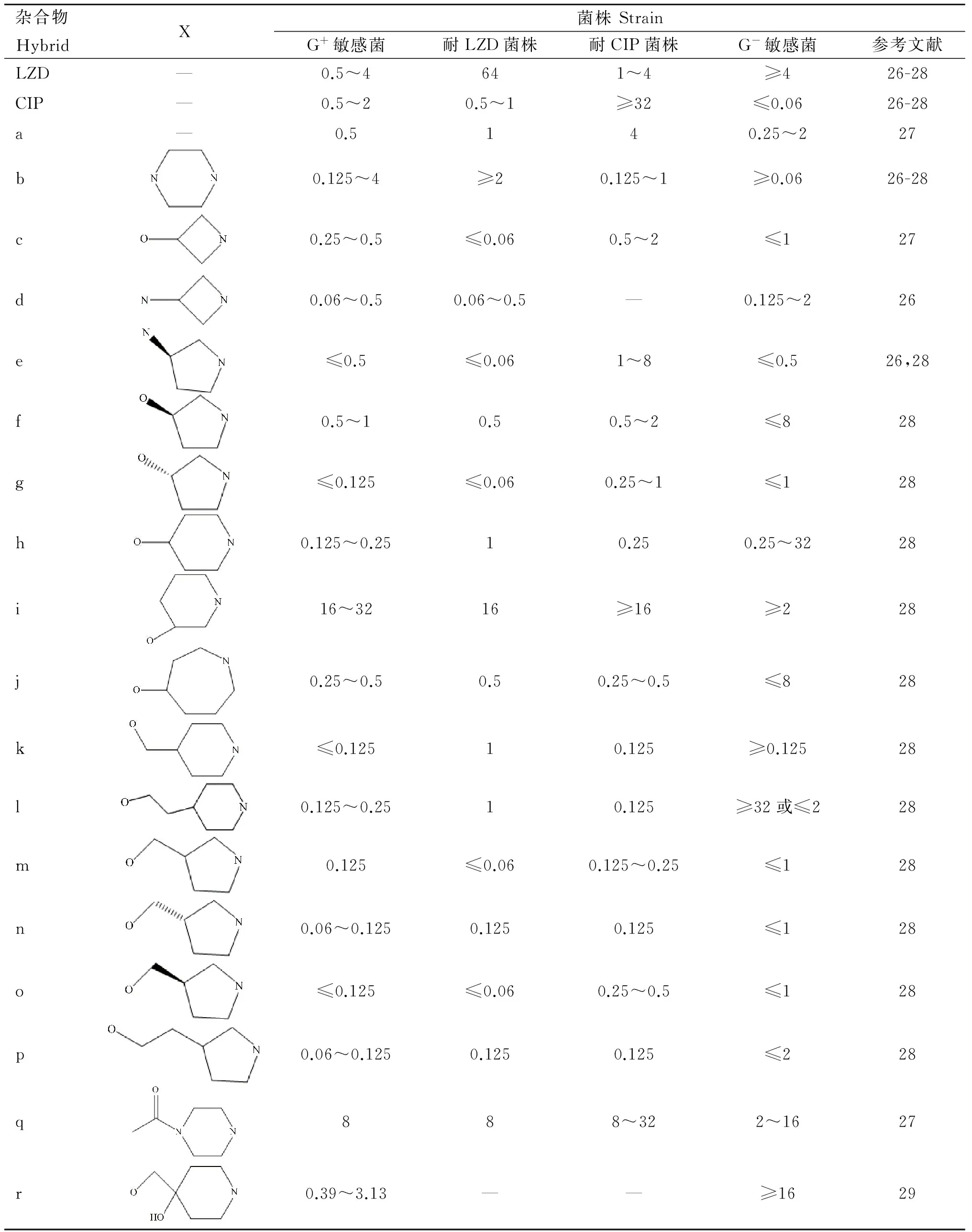
Table 1 Antibacterial activities of oxazolidinon-equinolone hybrids (MICs) μg·mL-1
“—”表示无/未评价
“—” means no datum/no evaluation
本实验室也基于噁唑烷酮—喹诺酮杂合物Ⅰ的拼合方式进行了相关研究,获得两个活性较优的化合物。以利奈唑胺、环丙沙星、恩诺沙星为对照药的生物活性评价表明两个化合物具有很好的抗菌活性(MICs≤0.25 μg·mL-1),抗菌谱与噁唑烷酮类抗生素更为相似,且效果明显优于利奈唑胺,对部分G-菌也很敏感(未发表资料)。
此外,T·卡布思纳等以杂合物1(图7)为例,研究噁唑烷酮—喹诺酮杂合物与其他抗生素的联合抗菌活性,结果表明当杂合物1与黏菌素联用时,对G-菌的活性显著增强,扩展了其抗菌谱,与β-内酰胺类或莫西沙星和利奈唑胺联用,出现高度协同作用,并可能恢复β-内酰胺类或莫西沙星和利奈唑胺对耐药菌株的活性[29]。

图7 噁唑烷酮—喹诺酮杂合物1的结构式Fig.7 The chemical structure of oxazolidinone-quinolone hybrids 1
在对噁唑烷酮—喹诺酮杂合物Ⅱ的体外抗菌活性评价中,选择3个杂合物对金黄色葡萄球菌、肺炎链球菌、黏膜炎莫拉菌的活性进行考察,结果MIC均≤0.063 μg·mL-1 [30]。
3.3 临床研究阶段的噁唑烷酮—喹诺酮杂合化合物——咔哒唑胺
咔哒唑胺(cadazolid)是由瑞士Actelion 公司开发,应用于艰难梭菌相关性腹泻(CDAD)的新型抗菌药,结构中同时包含噁唑烷酮和氟喹诺酮部分(图8),目前处于Ⅲ期临床研究阶段[31]。
3.3.1 咔哒唑胺作用机制 咔哒唑胺的作用机制主要是抑制细菌蛋白质的合成,而对DNA回旋酶/拓扑异构酶的抑制作用微弱。同时,氟喹诺酮部分为咔哒唑胺带来了独特的理化性质,使咔哒唑胺更容易渗透到细菌细胞中[32]。

图8 咔哒唑胺的结构式Fig.8 The chemical structure of cadazolid
3.3.2 咔哒唑胺抗菌活性及耐药性的产生 多项研究均已表明咔哒唑胺对艰难梭菌有很强的抑制作用。C. H. Chilton等以甲硝唑、万古霉素、莫西沙星及利奈唑胺为对照药物,考察了咔哒唑胺对100株艰难梭菌菌株(包括对甲硝唑敏感性降低的菌株、利奈唑胺耐药株、莫西沙星耐药株)的最小抑菌浓度(MIC),结果表明,咔哒唑胺对所有艰难梭菌菌株均敏感(MIC90为0.125 mg·L-1,MIC范围为0.03~0.25 mg·L-1),敏感性明显高于对照药物,而且咔哒唑胺对艰难梭菌的芽胞及毒素的产生也有较好的抑制作用[33]。M. U. Rashid等在评价咔哒唑胺对133株艰难梭菌菌株的体外活性以及后续对114株艰难梭菌株的研究中也发现了类似的结果[34-35]。此外,H. H. Locher等还以仓鼠和小鼠的艰难梭菌感染性腹泻为模型对咔哒唑胺在体内的抗菌活性进行了评估,结果表明,咔哒唑胺对模型鼠疗效确切,呈现剂量相关性,动物未出现死亡情况,与万古霉素的作用相当[36]。
尽管咔哒唑胺对G+菌(如金黄色葡萄球菌和肠球菌)有良好的抗菌活性,但对大多数G-菌活性较弱或几乎没有活性[32]。同时,咔哒唑胺对正常的肠道菌群影响很小,除双歧杆菌外,脆弱拟杆菌和乳酸菌均不受影响[33]。
艰难梭菌发生对于咔哒唑胺耐药的自发突变率很低(<10-10),明显低于莫西沙星和非达霉素(10-7~10-8),同时在连续传代试验中,咔哒唑胺的MICs值也没有显著增加,且咔哒唑胺对氟喹诺酮耐药株和利奈唑胺耐药株均有较好的活性,无交叉耐药性[32]。
3.3.3 咔哒唑胺药效学及安全性 咔哒唑胺的Ⅱ期临床试验招募了84例艰难梭菌感染患者,采用多中心、随机、双盲对照试验,以万古霉素(125 mg,po,qid)为对照,评估了3个剂量组(250、500、1 000 mg,po,bid)咔哒唑胺的有效性,服药周期为10 d。结果表明,各剂量组咔哒唑胺的疗效相似,无剂量相关性,且治愈率和维持治愈率均优于万古霉素[37]。咔哒唑胺的Ⅰ期临床试验和Ⅱ期临床试验均表明其安全性和耐受性良好,最常见的不良反应是头痛,同时有轻微的消化不良、眩晕等症状[31, 37]。
3.3.4 咔哒唑胺药动学及代谢特点 咔哒唑胺口服生物利用度极低,单剂量(30、100、300、1 000、3 000 mg)或多剂量(300、1 000、3 000 mg,po,bid,为期10 d)空腹给予健康受试者后,血药浓度2~3 h达峰,单剂量不超过3.3 ng·mL-1,多剂量不超过6.9 ng·mL-1,t1/2在13.02~14.19 h 之间,进食可增加Cmax,单剂量(300 mg)给药时,Cmax由0.73 ng·mL-1增至1.87 ng·mL-1 [38]。单剂量(3 000 mg)给予艰难梭菌感染患者,峰浓度为2.64 ng·mL-1 [38]。咔哒唑胺口服后主要以原型的形式经粪便排出,尿液中的药物原型很少[38-39]。
4 展 望
将作用于两个不同靶点的药效团拼合到一个稳定的分子中,并实现对两个不同靶点的双重作用,存在相当大的难度,有许多关键的问题需要考虑,包括拼合的方式、拼合分子的稳定性、拼合后分子理化性质及空间结构的变化等,这些问题对于拼合抗菌剂有效地发挥抗菌作用至关重要。
尽管如此,研究人员已经在噁唑烷酮和喹诺酮SAR相互兼容的条件下设计了一系列的噁唑烷酮—喹诺酮杂合抗菌剂,其体外抗菌谱包括各种临床相关易感以及抗性(如MRSA、QRSA、VRE和PRSP)G+菌和G-菌。而且从咔哒唑胺在临床上取得的进展也可以看出,噁唑烷酮和喹诺酮杂合物作为一种高效且低耐药倾向的新型抗菌剂化合物很有发展前景,值得进一步研究。
[1] BAUER P R, SAMPATHKUMAR P. Methicillin-resistantStaphylococcusaureusinfection in ICU: What is the best prevention strategy? [J].CritCareMed, 2017, 45(8): 1413-1414.
[2] ZAJONZ D, WUTHE L, RODLOFF A C, usw. Infektionen von Hüft- und Knieendoprothesen [J].DerChirurg, 2016, 87(4): 332-339.
ZAJONZ D, WUTHE L, RODLOFF A C, et al. Infections of the hip and knieendoprothesen[J].TheSurgeon, 2016, 87(4): 332-339.(in German)
[3] OKADA T, SATO Y, TOYONAGA Y, et al. Nationwide survey ofStreptococcuspneumoniaedrug resistance in the pediatric field in Japan[J].PediatrInt, 2016, 58(3): 192-201.
[4] MILLER W R, MURRAY B E, RICE L B, et al. Vancomycin-resistant enterococci: therapeutic challenges in the 21st century[J].InfectDisClinNorthAm, 2016, 30(2): 415-439.
[5] POKROVSKAYA V, BAASOV T. Dual-acting hybrid antibiotics: a promising strategy to combat bacterial resistance[J].ExpertOpinDrugDiscov, 2010, 5(9): 883-902.
[6] DALY J S, ELIOPOULOS G M, REISZNER E, et al. Activity and mechanism of action of DuP 105 and DuP 721, new oxazolidinone compounds[J].JAntimicrobChemother, 1988, 21(6): 721-730.
[7] YAGI B H, ZURENKO G E.Invitroactivity of linezolid and eperezolid, two novel oxazolidinone antimicrobial agents, against anaerobic bacteria[J].Anaerobe, 1997, 3(5): 301-306.
[8] SENIOR K. FDA approves first drug in new class of antibiotics[J].Lancet, 2000, 355(9214): 1523.
[9] IM W B, CHOI S H, PARK J Y, et al. Discovery of torezolid as a novel 5-hydroxymethyl-oxazolidinone antibacterial agent[J].EurJMedChem, 2011, 46(4): 1027-1039.
[10] KLUPP E M, BOTH A, BELMAR CAMPOS C, et al. Tedizolid susceptibility in linezolid-and vancomycin-resistantEnterococcusfaeciumisolates[J].EurJClinMicrobiolInfectDis, 2016, 35(12): 1957-1961.
[11] HOWE R A, WOOTTON M, NOEL A R, et al. Activity of AZD2563, a novel oxazolidinone, againstStaphylococcusaureusstrains with reduced susceptibility to vancomycin or linezolid[J].AntimicrobAgentsChemother, 2003, 47(11): 3651-3652.
[12] EDNIE L M, RATTAN A, JACOBS M R, et al. Antianaerobe activity of RBX 7644 (ranbezolid), a new oxazolidinone, compared with those of eight other agents[J].AntimicrobAgentsChemother, 2003, 47(3): 1143-1147.
[13] GORDEEV M F, LUEHR G W, PATEL D V, et al. Preparation ofN-acyl-3-aryl-2-oxooxazolidine-5-methanammines as bactericides: WO, 2001009107[P]. 2000-07-26.
[14] JADHAVAR P S, VAJA M D, DHAMELIYA T M, et al. Oxazolidinones as anti-tubercular agents: discovery, development and future perspectives[J].CurrMedChem, 2015, 22(38): 4379-4397.
[15] BARBACHYN M R, FORD C W. Oxazolidinone structure-activity relationships leading to linezolid[J].AngewChemIntEdEngl, 2003, 42(18): 2010-2023.
[16] DAS B, RUDRA S, YADAV A, et al. Synthesis and SAR of novel oxazolidinones: discovery of ranbezolid[J].BioorgMedChemLett, 2005, 15(19): 4261-4267.
[17] XIN Q S, FAN H X, GUO B, et al. Design, synthesis, and structure-activity relationship studies of highly potent novel benzoxazinyl-oxazolidinone antibacterial agents[J].JMedChem, 2011, 54(21): 7493-7502.
[18] LESHER G Y, FROELICH E J, GRUETT M D, et al. 1,8-naphthyridine derivatives. A new class of chemotherapeutic agents[J].JMedPharmChem, 1962, 91: 1063-1065.
[19] CORREIA S, POETA P, HÉBRAUD M, et al. Mechanisms of quinolone action and resistance: where do we stand?[J].JMedMicrobiol, 2017, 66(5): 551-559.
[20] CROSS R M, MONASTYRSKYI A, MUTKA T S, et al. Endochin optimization: structure-activity and structure-property relationship studies of 3-substituted 2-methyl-4(1H)-quinolones with antimalarial activity[J].JMedChem, 2010, 53(19): 7076-7094.
[21] MAIGNAN J R, LICHOROWIC C L, GIARRUSSO J, et al. ICI 56,780 Optimization: Structure-activity relationship studies of 7-(2-phenoxyethoxy)-4(1H)-quinolones with antimalarial activity[J].JMedChem, 2016, 59(14): 6943-6960.
[22] OUAHROUCH A, IGHACHANE H, TAOURIRTE M, et al. Benzimidazole-1,2,3-triazole hybrid molecules: synthesis and evaluation for antibacterial/antifungal activity[J].ArchPharm(Weinheim), 2014, 347(10): 748-755.
[23] KAROLI T, MAMIDYALA S K, ZUEGG J, et al. Structure aided design of chimeric antibiotics[J].BioorgMedChemLett, 2012, 22(7): 2428-2433.
[24] ROBERTSON G T, BONVENTRE E J, DOYLE T B, et al.Invitroevaluation of CBR-2092, a novel rifamycin-quinolone hybrid antibiotic: microbiology profiling studies withstaphylococciandstreptococci[J].AntimicrobAgentsChemother, 2008, 52(7): 2324-2334.
[25] POKROVSKAYA V, BELAKHOV V, HAINRICHSON M, et al. Design, synthesis, and evaluation of novel fluoroquinolone-aminoglycoside hybrid antibiotics[J].JMedChem, 2009, 52(8): 2243-2254.
[26] HUBSCHWERLEN C, SPECKLIN J L, SIGWALT C, et al. Design, synthesis and biological evaluation of oxazolidinone-quinolone hybrids[J].BioorgMedChem, 2003, 11(10): 2313-2319.
[27] GORDEEV M F, HACKBARTH C, BARBACHYN M R, et al. Novel oxazolidinone-quinolone hybrid antimicrobials[J].BioorgMedChemLett, 2003, 13(23): 4213-4216.
[28] HUBSCHWERLEN C, SPECKLIN J L, BAESCHLIN D K, et al. Structure-activity relationship in the oxazolidinone-quinolone hybrid series: influence of the central spacer on the antibacterial activity and the mode of action[J].BioorgMedChemLett, 2003, 13(23): 4229-4233.
[29] T·卡布思纳, A·达尔霍夫. 含噁唑烷酮—喹诺酮的联合疗法在治疗细菌感染中的应用: 中国, CN105246476A[P]. 2016-01-13.
KAPSNER T, DALHOFF A. Combination therapy comprising oxazolidinone-quinolones for use in treating bacterial infections:China, CN105246476A[P]. 2016-01-13. (in Chinese)
[30] 克里斯蒂安·哈伯舒兰, 菲利普·潘彻德, 克里斯汀·西格瓦尔特,等.作为抗菌化合物的恶唑烷酮—喹诺酮混合物: 中国,CN101238135B[P]. 2011-05-11.
PANCHAUD P, SIGWALT C, HUBSCHWERLEN C, et al. Oxazolidinone-quinolone hybrids as antibacterial compounds: China, CN101238135B[P]. 2011-05-11. (in Chinese)
[31] ENDRES B T, BASSèRES E, ALAM M J, et al. Cadazolid for the treatment ofClostridiumdifficile[J].ExpertOpinInvestigDrugs, 2017, 26(4): 509-514.
[32] LOCHER H H, CASPERS P, BRUYRE T, et al. Investigations of the mode of action and resistance development of cadazolid, a new antibiotic for treatment ofClostridiumdifficileinfections[J].AntimicrobAgentsChemother, 2014, 58(2): 901-908.
[33] CHILTON C H, CROWTHER G S, BAINES S D, et al.Invitroactivity of cadazolid against clinically relevantClostridiumdifficileisolates and in aninvitrogut model ofC.difficileinfection[J].JAntimicrobChemother, 2014, 69(3): 697-705.
[34] RASHID M U, LOZANO H M, WEINTRAUB A, et al.Invitroactivity of cadazolid againstClostridiumdifficilestrains isolated from primary and recurrent infections in Stockholm, Sweden[J].Anaerobe, 2013, 20: 32-35.
[35] RASHID M U, DALHOFF A, WEINTRAUB A, et al.Invitroactivity of MCB3681 againstClostridiumdifficilestrains[J].Anaerobe, 2014, 28: 216-219.
[36] LOCHER H H, SEILER P, CHEN X H, et al.Invitroandinvivoantibacterial evaluation of cadazolid, a new antibiotic for treatment ofClostridiumdifficileinfections[J].AntimicrobAgentsChemother, 2014, 58(2): 892-900.
[37] LOUIE T, NORD C E, TALBOT G H, et al. Multicenter, double-blind, randomized, phase 2 study evaluating the novel antibiotic cadazolid in patients withClostridiumdifficileinfection[J].AntimicrobAgentsChemother, 2015, 59(10): 6266-6273.
[38] BALDONI D, GUTIERREZ M, TIMMER W, et al. Cadazolid, a novel antibiotic with potent activity againstClostridiumdifficile: safety, tolerability and pharmacokinetics in healthy subjects following single and multiple oral doses[J].JAntimicrobChemother, 2014, 69(3): 706-714.
[39] GEHIN M, DESNICA B, DINGEMANSE J. Minimal systemic and high faecal exposure to cadazolid in patients with severeClostridiumdifficileinfection[J].IntJAntimicrobAgents, 2015, 46(5): 576-581.
