黄芪多糖通过抑制NF-κB和JNK信号通路减轻LPS诱导的小鼠心肌细胞凋亡
2018-01-24王洪新鲁美丽
韩 琳,王洪新,鲁美丽
(锦州医科大学心脑血管药物研究重点实验室,辽宁 锦州 121001)
心肌细胞凋亡是指心肌细胞程序性自主死亡。近年研究发现,氧化应激、缺血/再灌损伤等可诱导细胞凋亡[1-2],在各种心血管疾病的发病机制和预后中发挥重要作用。所以,抗心肌细胞凋亡对心脏疾病的治疗是一个重要的靶点。促分裂原激活蛋白激酶(mitogen activated protein kinases,MAPKs)通路是真核细胞调控介导细胞内信号转导的重要系统,参与动物细胞的增殖、凋亡、分化、转化等生物学反应,包括ERK、JNK、p38 MAPK 以及 ERK5/BMK1四条途径[3]。通常情况下,ERK起保护细胞作用,而JNK和p38 MAPK则表现为促凋亡作用[4]。当JNK被不同的刺激因子激活后,其转移至细胞核内,激活其下游作用底物——核内转录因子(c-Jun),并磷酸化。研究发现,c-Jun大量磷酸化可导致细胞凋亡和分化[5]。ASK1-MKK4/7-JNK信号通路可以激活细胞氧化应激通路,调控NF-κB的活性,使一氧化氮(NO)的生成增加,诱导细胞损伤[6]。NF-κB是核转录因子,通过调节炎症因子、诱导酶、趋化因子等,在免疫、炎症、凋亡中发挥重要作用[7]。NF-κB既可调节其下游的促炎症因子肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)、白细胞介素-1β(interleukin-1β,IL-1β)的表达,又可被其反馈激活,进而放大炎症反应,上调NF-κB的表达,诱导细胞凋亡[8]。
黄芪多糖(Astragalus polysaccharide,APS)是黄芪的主要活性成分之一,研究发现,APS具有免疫促进剂或调节剂及增强机体抵抗力的作用,尤其具有保护心肌的作用[9]。现已证实,脂多糖(lipopolysaccharide, LPS)能刺激NF-κB和JNK信号通路的活化[10],且本实验室前期实验已证明APS可抑制心肌细胞凋亡[11],但其具体机制尚不明确。本研究采用LPS诱导小鼠心肌细胞凋亡,通过检测相关凋亡因子表达水平及其在心肌组织中的浸润情况,阐明APS通过抑制NF-κB和JNK信号通路减轻LPS诱导的小鼠心肌细胞凋亡。
1 材料与方法
1.1药品与试剂黄芪多糖,南京景竹生物技术有限公司,批号JZ150206A,APS纯度为98%;LPS,购自北京鼎国昌盛生物技术有限公司;TUNEL试剂盒购自Roche公司;IL-β、TNF-α ELISA试剂盒购自R&D公司;一抗p-JNK、JNK、IκB-α、Bcl-2、caspase-3、β-actin均购自ABclonal公司;Bax抗体购自Proteintech公司;HRP山羊抗兔IgG(H+L)、NF-κB购自北京博奥森生物技术有限公司;辣根过氧化物酶标记山羊抗小鼠IgG(H+L)、ECL化学发光显色试剂盒、BCA试剂盒、细胞质细胞核蛋白提取试剂盒、FITC标记山羊抗兔IgG(H+L),均购自北京鼎国昌盛生物技术有限公司;胰蛋白酶、二甲基亚砜(dimethyl sulfoxide,DMSO)均购自美国 Sigma公司;高糖DMEM培养基购自Gibco公司;小牛血清购自美国 Hyclone公司。
1.2实验动物与细胞♂ SPF级昆明小鼠40只,7周龄,体质量(20±2)g,由锦州医科大学实验动物中心提供,动物合格证号:SCXK2014-0004。H9c2细胞株购自武汉博士德生物有限公司。
1.3仪器DMI 3000B 倒置显微镜(德国Leica公司);凝胶成像仪、电泳仪、电泳槽、转印机(美国Bio-Rad公司);酶标仪(上海赛默飞世尔仪器有限公司);小动物超声影像系统Prospect3.0(台湾S-Sharp公司);超净工作台(江苏吴县市净化技术研究所);CO2孵箱(美国Sheldon Manufacturing Inc公司)。
1.4方法
1.4.1动物实验分组 SPF级昆明小鼠40只,随机分为空白对照组、模型组、APS(200、400、800 mg·kg-1)组,每组8只,对照组和模型组给予同等剂量的生理盐水灌胃。14 d后,给药组和模型组腹腔注射LPS(10 mg·kg-1),8 h后进行下一步实验。
1.4.2H9c2心肌细胞的培养 用含15%胎牛血清的高糖DMEM培养液,于37℃、5% CO2恒温培养箱中进行常规培养。至密度90%左右时传代,以2×107·L-1浓度接种6孔板,约48 h后换液1%胎牛血清16 h,进行诱导分化后开始实验。实验所需药品APS为水溶性,以三蒸水溶解后,用0.22 μm针式滤器过滤除菌;APS预先孵育30 min后,加入1 mg·L-1LPS,24 h后进行各指标的测定。实验共分为5组:空白对照组、LPS模型组(1 mg·L-1)、APS低、中、高剂量组(10、25、50 mg·L-1)。
1.4.3超声心动检测心脏情况 对照组和模型组给予同等剂量的生理盐水灌胃。14 d后,给药组和模型组腹腔注射脂多糖10 mg·kg-1,8 h后,小鼠吸入麻醉,超声心动测左心室射血分数(ejection fraction,EF)、左心室缩短分数(fractional shortening,FS)、搏出量(stroke volume,SV)、心输出量(cardiac output,CO)。
1.4.4ELISA法检测IL-1β、TNF-α水平 小鼠摘眼球取血于EP管中,常温下静置2 h,析出血清后,12 000 r·min-1离心10 min,取上清液。根据试剂盒说明书操作,酶标495 nm波长处测定吸光度(OD)值。
1.4.5TUNEL染色检测心肌细胞凋亡 取心肌组织常规固定,冰冻切片,用PBS冲洗30 min,甩干后3%过氧化氢-甲醇处理10 min,PBS浸洗3次,每次5 min,然后在冰上进行如下操作:0.1% Triton X-100、0.1%柠檬酸钠溶液处理2 min,PBS浸洗2次,每次5 min,TUNEL反应混合液37℃孵育1 h,PBS浸洗3次。吸净PBS,用荧光显微镜进行观察。
1.4.6细胞核细胞质蛋白的提取 将心肌组织碾碎成细小碎片,按照20 ∶1的比例混合细胞质蛋白抽提试剂A和B,加入PMSF配制成终浓度为1 mmol·L-1的混合液,每60 mg的组织中加入200 μL匀质液,冰浴放置15 min,15 000 r·min-1离心5 min,把上清移至EP管中,此为细胞质蛋白。每20 μL的细胞沉淀再加入200 μL细胞质蛋白抽提试剂A,超声10 s,冰浴15 min,加入细胞质蛋白抽提试剂B,超声10 s,冰浴1 min,再超声10 s,4℃、15 000 r·min-1离心5 min,吸取上清液,此为细胞质蛋白。然后将沉淀加入细胞核蛋白抽提剂,最高涡速30 s,冰浴2 min,再次涡速30 s,共30 min,然后4℃、15 000 r·min-1离心10 min,取上清液,此为细胞核蛋白。贴壁细胞用细胞刮子刮下并用移液器吹打,离心收集,然后实验方法与上述相同。
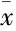
Tab 1 Effects of APS on LPS-induced hemodynamics in mice(±s,n=8)
EF: Left ventricular ejection fraction; FS: Left ventricular shortening score; SV: Stroke volume; CO: Cardiac output.**P<0.01vscontrol;##P<0.01vsLPS model
1.4.7Western blot检测 取冷冻心肌组织100 mg和心肌细胞悬液200 μL,加入1 mL蛋白裂解液,用超声细胞破碎仪于冰上超声,4℃、12 000 r·min-1离心10 min,将离心后的上清BCA法测定蛋白含量。SDS-PAGE凝胶电泳,将蛋白质转移到硝酸纤维素滤膜上。10%的BSA室温封闭2 h后,分别加IκB-α(1 ∶1 000)、NF-κB (1 ∶500)、p-JNK(1 ∶1 000)、JNK(1 ∶1 000)、Bax(1 ∶5 000)、Bcl-2(1 ∶1 000)、caspase-3(1 ∶1 000)、 Lamin-B(1 ∶1 000)、β-actin(1 ∶5 000)一抗,孵育过夜。然后加入HRP标记的羊抗小鼠二抗(1 ∶5 000),室温孵育l.5 h,ECL发光剂显色发光,生物发光成像分析仪检测分析。

2 结果
2.1APS对小鼠心功能的影响超声心动图结果显示,不同浓度的APS能有效地防止LPS引起的心肌功能的减退,其作用存在剂量依赖性。如Tab 1所示,与正常组相比,模型组的EF和FS都明显降低,模型组心肌收缩功能明显改变(P<0.01);与模型组相比,APS(400、800 mg·kg-1)处理组EF和FS明显恢复(P<0.01)。
2.2APS对血清TNF-α、IL-1β水平的影响TNF-α、IL-1β是促炎症因子,它们可以被LPS诱导,并激活NF-κB和JNK信号通路,进而引起心肌细胞凋亡。Tab 2的ELISA结果表明,LPS诱导后TNF-α、IL-1β水平明显升高(P<0.01),APS (400、800 mg·kg-1)能有效抑制LPS刺激后TNF-α、IL-1β水平的升高,其作用呈剂量依赖性(P<0.01)。
2.3APS对心肌细胞凋亡的影响如Fig 1所示,与正常对照组相比,LPS组细胞凋亡明显增加(P<0.01)。与LPS组相比,APS (400、800 mg·kg-1)干预后能明显降低凋亡(P<0.01),而APS 200 mg·kg-1组的凋亡率与LPS组比较无差别。
Fig 1 Effect of APS on cardiomyocyte apoptosis(×200)
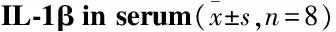
Tab 2 Effects of APS on contents of TNF-α and
**P<0.01vscontrol;##P<0.01vsLPS model
2.4APS对LPS诱导的JNK通路的影响APS是否通过JNK信号通路而抑制心肌细胞凋亡呢?在体内与体外实验中,与正常对照组相比,LPS组p-JNK明显升高(P<0.01);与LPS组相比,APS中、高剂量组p-JNK明显降低(P<0.01),且呈剂量依赖性,而JNK则没有明显变化(Fig 2)。
2.5APS对LPS诱导的NF-κB信号通路的影响Fig 3 Western blot结果表明,在体内与体外实验中,LPS处理后细胞质中的IκB-α、NF-κB蛋白表达降低(P<0.01),而细胞核中NF-κB的表达增高(P<0.01); APS中、高剂量可明显抑制IκB-α降解和NF-κB向核内转移(P<0.05)。提示APS对LPS诱导的IκB-α的降解和NF-κB向核内转移有抑制作用。
2.6APS对Bcl-2、caspase家族的影响Fig 4结果显示,与对照组相比,模型组Bax、caspase-3蛋白表达明显增高,而Bcl-2表达降低(P<0.01);APS中、高剂量组中Bax、caspase-3的蛋白表达明显降低,Bcl-2表达增高(P<0.01)。提示APS对caspase家族、Bax蛋白表达有抑制作用,而促进Bcl-2蛋白表达。
3 讨论
心肌细胞凋亡与许多心血管疾病的发生、发展有关联。Bcl-2特异性抑制细胞凋亡的主要机制是其在线粒体水平上与Bid、Bim或Bad结合,却与Bax或Bak解离,从而维护线粒体膜的完整性,因而防止膜间隙中凋亡蛋白的渗出;同时还可以抑制Ca2+的释放,降低线粒体对Ca2+的摄取,从而抑制心肌细胞凋亡。另外,Bcl-2还能直接与凋亡蛋白活性因子-1(Apaf-1)结合,形成Bcl-2/ Apaf-1/caspase-9复合物,也能阻断caspase的始动激活[12]。caspase-3是caspase家族在心肌细胞凋亡过程中重要的蛋白酶、多种凋亡途径的共同下游效应因子、心肌细胞凋亡蛋白酶级联反应的必经之路,可直接诱导细胞凋亡。本研究中,LPS模型组心肌组织中Bcl-2降低, Bax、caspase-3的蛋白表达增加。而APS中、高剂量组均可增加Bcl-2表达,降低Bax、caspase-3的蛋白表达。表明APS可以对抗LPS诱导的心肌细胞凋亡,对心脏起保护作用。
Fig 2 Effect of APS on LPS-induced JNK pathway in vivo and in vitro(±s,n=8)
A: The protein levels of JNK and p-JNK were measured by Western blotinvivoexperiments; B: The protein levels of JNK and p-JNK were measured by Western blotinvitroexperiments.**P<0.01vscontrol;##P<0.01vsLPS model
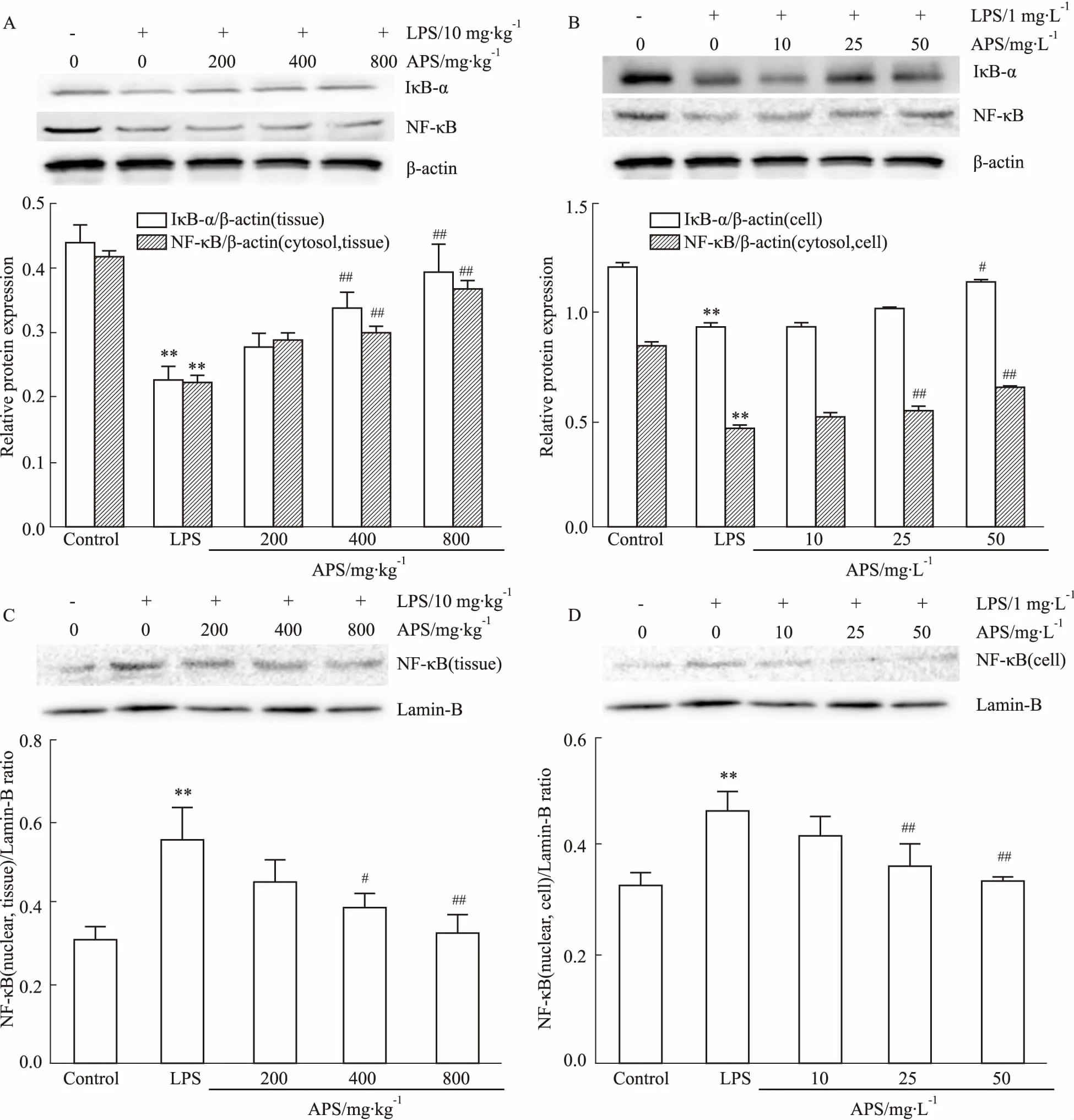
Fig 3 Effect of APS on LPS-induced NF-κB signaling pathway in vivo and in vitro(±s,n=8 )
The contents of IκB-α and NF-κB protein in cytoplasm of cardiomyocyte induced by LPS-induced myocardiuminvivo(A) andinvitro(B); APS inhibited the content of NF-κB protein in LPS-induced nuclearinvivo(C) andinvitro(D).**P<0.01vscontrol;#P<0.05,##P<0.01vsLPS model.
既往研究显示,NF-κB和JNK信号通路可以诱导心肌细胞凋亡[13-14]。c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)是MAPKs家族成员,在细胞凋亡中也是重要的信号通路,当其受到外界刺激后,JNKK1/MKK4/SEK1或者JNKK2/MKK7介导JNK上Thr183和Tyr185位点磷酸化,使JNK完全活化并具有酶催化活性[7]。而当JNK被活化后,其转移到细胞核内,并使它主要的下游作用物c-Jun磷酸化,激活依赖于转录的凋亡信号通路[15]。p-JNK诱导心肌细胞凋亡可能的机制是既可促进多种凋亡蛋白的表达,又可通过线粒体水平发挥其有效抗凋亡作用,还可作用于Bcl-2家族中促凋亡蛋白Bax、Bak等,诱导细胞色素释放入细胞质内,从而激活细胞凋亡的起始[12]。本实验体内、体外研究中,APS中、高剂量组可以明显降低JNK蛋白的磷酸化,表明APS对抗LPS诱导的心肌细胞凋亡可能与JNK信号通路有关。

Fig 4 Effects of APS on Bcl-2 and caspase
**P<0.01vscontrol;##P<0.01vsLPS model
NF-κB是一个转录因子,在炎症、凋亡等细胞生理活动中发挥重要作用。NF-κB的活化可被炎症因子IL-1β、TNF-α激活,进而引起一系列的心肌损伤。NF-κB既可以调节IL-1β、TNF-α等炎症因子的释放,又可以被其反馈调节。IκB是一个分子复合物,当IκB被激活磷酸化后,NF-κB p65/50异源二聚体从复合物IκB中脱落,并从细胞质进入细胞核,诱导细胞凋亡。我们研究发现,APS中、高剂量组可以有效抑制LPS诱导的IκB磷酸化和随后的降解,而且阻止NF-κB p65/50进入细胞核,并使其下游细胞因子IL-1β、TNF-α释放减少。所以我们推断,APS可能通过抑制信号通路NF-κB来抑制LPS诱导的心肌细胞凋亡。
综上所述,LPS通过活化NF-κB和JNK信号通路诱导心肌细胞凋亡,APS减少LPS所致心肌细胞凋亡的机制可能与抑制NF-κB和JNK信号通路有关。本实验部分揭示了APS的作用机制,并且为心血管疾病的治疗提供了新的靶点,为寻找新的治疗药物提供了新的思路。
(致谢: 本实验完成于锦州医科大学心脑血管药物研究重点实验室,实验过程中得到该实验室全体老师的指导及同学的协助,在此表示感谢。)
[1] Vila-Petroff M,Salas M A,Said M,et al. CaMK Ⅱ inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury[J].CardiovascRes,2007,73(4):689-98.
[2] 陈丹丹,彭 成,万 峰, 等. 氢溴酸樟柳碱对抗大鼠急性脑缺血/再灌注损伤的作用机制研究[J]. 中国药理学通报,2017,33(8):1096-102.
[2] Cheng D D, Peng C, Wan F, et al. The protective mechanism of anisodine hydrobromide gainstcerebral ischemia-reperfusion injury in rats[J].ChinPharmacolBull, 2017,33(8):1096-102.
[3] 叶 蕾,陈芝芸,严茂祥,等.肝纤维化大鼠肝组织MAPKs信号通路的活化及意义[J]. 中华中医药学刊,2013,31(12):2748-50.
[3] Ye L, Chen Z Y, Yan M X, et al. Activation and significance of MAPKs signaling pathway in hepatic tissue of rats with hepatic fibrosis [J].ChinJTraditChinMed, 2013,31(12):2748-50.
[4] Patterson K I, Brummer T, O’Brien P M, et al, Dual-specificity phosphatases: critical, regulators with diverse cellular targerts [J].BiochemJ,2009,418(3):475-89.
[5] Weiss C, Schneider S, Wagner E F, et al. JNK phosphorylation relieves HDAC3-dependent suppression of the transcriptional activity of c-jun[J].EMBOJ,2003,22(14):3686-95.
[6] Su C C, Chen J Y, Din Z H, et al. 13-Acetoxysarcocrassolide induces apoptosis on human gastric carcinoma cells through mitochondria-related apoptotic pathways:p38/JNK activation and PI3K/AKT suppression[J].MarDrugs,2014,12(10): 5295-315.
[7] Lawrence T, Willoughby D A, Gilroy D W. Anti-inflammatory lipid mediators and insights into the resolution of inflammation[J].NatRevImmunol, 2002,2(10):787-95.
[8] Yeom M, Kim J H, Min J H, et al. Xanthii fructus inhibits inflammatory responses in LPS-stimulated RAW 264.7 macrophages through suppressing NF-κB and JNK/p38MAPK[J].JEthnopharmacol, 2015,176:394-401.
[9] 柏冬志,东 方,唐文婷, 等.黄芪多糖药理作用的研究进展[J]. 黑龙江医药,2014,27(1):103-6.
[9] Bai D Z,Dong F,Tang W T,et al. Research progress on pharmacological action of Astragalus polysaccharide[J].HeilongjiangMed,2014,27(1):103-6.
[10] 孙雪芳,王洪新,梁灵君,等.黄芪多糖通过TLR4/NF-κB信号通路抑制脂多糖诱导的大鼠心肌细胞肥大[J]. 中国药理学通报,2013,29(2):208-12.
[10] Sun X F, Wang H X, Liang L J, et al, Inhibition of Astragalus polysaccharide in lipopolysccharide-induced cardiomyocyte hypertrophy in rats through the TLR4/NF-κB signal transduction [J].ChinPharmacolBull, 2013,29(2):208-12.
[11] 于胜男,曹琼丹,鲁美丽, 等. 黄芪多糖对糖尿病大鼠心肌细胞凋亡的影响[J]. 中药药理与临床,2015,31(4):102-5.
[11] Yu S N,Cao Q D, Lu M L,et al, Effect of Astragalus polysaccharide on myocardial cell apoptosis in diabetic rats [J].PharmacolClinChinMaterMed,2015,31(4):102-5.
[12] Takagi Y, Nozaki K, Sugmo T, et al. Phosphorylation of c-Jun NH(2)-terminal kinase and p38 mitogen-activated protein kinase after transient forebrain ischemia in mice[J].NeurosciLett,2000,294(2):117-20.
[13] Paul A, Cuenda A, Bryant C E, et al. Involvement of mitogen-activated protein kinase homologues in the regulation of lipopolysaccharide-mediated induction of cyclo-oxygenase-2 but not nitric oxide synthase in RAW 264.7 macrophages [J].CellSignal, 1999,11(7):491-7.
[14] Uto T, Fujii M, Hou D X. 6-(Methylsulfinyl) hexyl isothiocyanate suppresses inducible nitric oxide synthase expression through the inhibition of Janus kinase 2-mediated JNK pathway in lipopolysaccharide-activated murine macrophages[J].BiochemPharmacol, 2005,70(8): 1211-21.
[15] 魏 娜,贺海波,张长城, 等.JNK信号通路与细胞凋亡关系的研究进展[J]. 中国临床药理学与治疗学,2013,18(7):807-12.
[15] Wei N, He H B, Zhang C C, et al. Advances in the relationship between JNK signaling pathway and apoptosis[J].ChinJClinPharmacolTher,2013,18(7):807-12.
