姜黄素环糊精分子包合物的构建和优化
2018-01-18赵春景张景勍
李 艺, 梅 虎, 赵春景, 王 红, 罗 文, 张景勍*
(1.重庆医科大学 药物高校工程研究中心,重庆 400016;2.重庆大学 生物工程学院,重庆 400023;3.重庆医科大学附属第二医院药剂科,重庆 400010)
姜黄素(curcumin,CCN)最早是从植物根茎提炼出来的一种印度香料,作为抗炎草药,具有悠久的使用历史。CCN溶于乙醇、丙酮和氯仿等有机溶剂,几乎不溶于水及乙醚。药理作用广泛,安全有效性高,价格低廉,使其具有广阔的应用前景。但由于CCN的溶解度极低,使其口服吸收不完全,代谢消除迅速,生物利用度低,限制了其应用。药物被环糊精大分子包合后可明显改善药物的理化性质,提高药物的水溶性和稳定性,提高药物的生物利用度以及改变药物在体内的分布情况等。为了改善CCN在体内的吸收,提高其口服生物利用度,本实验选用水溶性大、生物相容性好的的羟丙基-β-环糊精(hydroxypropyl beta cyclodextrin,HBCD) 为材料,构建了姜黄素羟丙基-β-环糊精分子包合物(curcumin-cyclodextrin inclusion complexes,CCIC),通过显微观察法、红外光谱法、差示量热扫描法验证了包合物的形成,并优化了CCN的制备工艺,成功提高了CCN在水溶液中的溶解度,以计算机模拟了CCIC的分子结构及CCN分子包合前后的能量变化。
1 材料与方法
1.1 材料和仪器
1.1.1 主要材料、试剂 羟丙基-β-环糊精:江苏泰兴新鑫医药辅料有限公司产品;姜黄素:陕西森弗生物制药有限责任公司产品;去离子水,其它试剂均为分析纯。
1.1.2 主要仪器 Monobloc AB204-E型电子天平:瑞士METTLER-TOLEDO仪器公司产品;UV-3150型紫外分光光度仪:日本岛津公司产品;DZF-6020型真空干燥箱:上海博运实业有限公司医疗设备厂制造;DF-101S型集热式恒温加热磁力搅拌器:巩义市于华仪器有限责任公司产品;UV-7504型紫外可见分光光度仪:上海精密科学仪器有限公司产品;光学显微镜:凤凰光学集团有限公司产品;STA 449C型综合热分析仪:德国耐驰仪器制造有限公司产品;WQF-510傅里叶变换红外光谱仪:上海第二光学仪器厂制造;THZ-82水浴恒温振荡器:金坛市荣华仪器制造有限公司产品。
1.2 实验方法
1.2.1 含量测定方法的确定 精密称取CCN 11.1 mg加入适量乙醇溶液溶解,将其移入100 mL的棕色容量瓶中,并用乙醇稀释至刻度,摇匀制成储备液备用。取储备液稀释适当倍数,以乙醇溶液为空白对照,在200~800 nm进行紫外扫描得CCN紫外吸收光谱,确定最大吸收波长。取HBCD适量溶于乙醇中,以乙醇溶液为空白对照,在200~800 nm进行紫外扫描,确定辅料有无干扰。精密量取上述母液10 mL于100 mL的容量瓶中,用无水乙醇定容至刻度线, 稀释摇匀。 取该溶液 1、2、3、4、5 mL于10 mL的容量瓶中,用无水乙醇定容至刻度线,稀释摇匀。以乙醇溶液为空白对照,在425 nm处测吸光度,以CCN质量浓度(C)对吸光度(A)进行线性回归,绘制标准曲线。按线性范围分别配制高、中、低3种浓度的CCN乙醇溶液,在425 nm测定吸光度,计算实际浓度。同日内测定日内精密度,连续3 d测定日间精密度。称取已知含量的CCIC适量溶于乙醇溶液中,移入10 mL的容量瓶加乙醇溶液至刻度。取1 mL上述溶液分别于9个10 mL的容量瓶中。取CCN储备液1.1、1.4、1.7 mL于上述容量瓶中,每浓度设置3组平行,用乙醇定容至刻度线摇匀。取出适量用乙醇稀释后,于425 nm处测定吸光度值,代入回归方程求出药物浓度,并计算回收率。
1.2.2 CCIC中CCN含量的测定 精密称取CCIC适量溶于乙醇溶液中,移至10 mL的容量瓶中,用乙醇稀释至刻度摇匀。取上述溶液再用乙醇溶液稀释适当倍数,于425 nm处测定吸光度,代入标准方程,计算CCIC中CCN的含量。
1.2.3 CCIC包合物的制备 采用研磨法制备CCIC,称取适量HBCD加入少量蒸馏水,研磨均匀,再缓慢加入CCN,边加边研磨至规定时间,适当干燥,沉淀用乙酸乙酯洗涤3次,再于30℃真空干燥3 h,研细即得 CCIC[1]。
1.2.4 CCIC最佳制备工艺的选择 选择B投料比(CCN与HPCD的摩尔比)、C包合温度、D研磨时间这3种对包合物制备影响较大的因素作为考察对象,每因素各取3水平,采用L9(34)正交实验表设计实验方案,见表1。
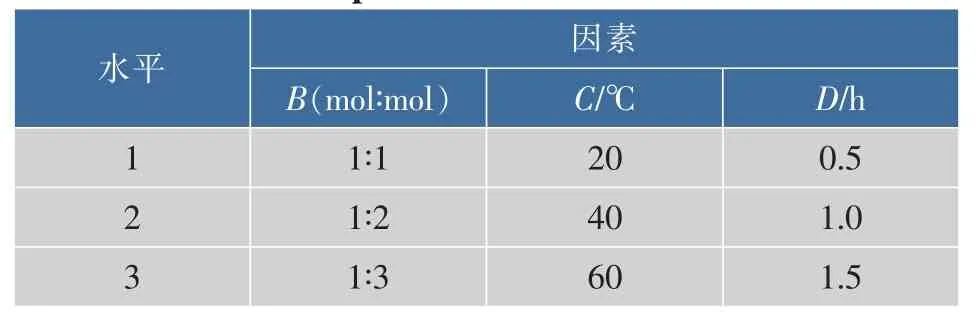
表1 CCIC制备工艺因素-水平表Table 1 Prepare of CCIC levels of factors
按正交实验设计表进行试验,以CCN溶解度为考察指标,确定最佳制备工艺[2]。
1.2.5 相溶解度法测定CCN与HBCD的包合常数精密称取 0.2、0.4、0.6、0.8、1.0 mmol HBCD 溶于5 mL蒸馏水中,向上述溶液中加入过量CCN,放入恒温振荡器中(25±2)℃,振荡24 h使溶解达到平衡状态。取出溶液1 mL,微孔滤膜过滤,滤液用乙醇稀释至适当倍数,在425 nm处测定紫外吸光度。以CCN含量对HBCD含量作图,即得相溶解度图[3-5]。
1.2.6 溶解度测定 称取适量CCN溶于25 mL蒸馏水中制备成过饱和溶液,于磁力搅拌器上搅拌,不同时间取样1 mL,用0.22 μm微孔滤膜过滤,滤液用乙醇稀释适当倍数,在425nm处测定吸光度值。溶解平衡时间为相邻样品测定的A值小于0.004时所对应的时间。按最优化处方工艺制备CCIC,称取适量溶于25 mL蒸馏水中制备成CCN过饱和溶液,同方法1.2.2取样并处理,测定吸光度值,计算得CCIC中CCN在蒸馏水中溶解度[6]。
1.2.7 包合物的鉴定 包合物的鉴定有显微观察法,差示量热扫描分析和红外分光光度法。显微观察法是分别取CCN、HBCD、CCN与HBCD物理混合物、CCIC在光学显微镜下观察。差示量热扫描分析是分别取CCN、HBCD、CCN与HBCD物理混合物、CCIC进行差示扫描量热分析,以Al2O3为参比,升温速度为10℃/min,氮气流速为20 mL/min,扫描范围为-20~350℃,取样量为5 mg。分别记录升温曲线。红外分光光度法是将CCN、HBCD、CCN与HBCD物理混合物、CCIC加入适量KBr压片,在4000~400 cm-1范围内累加扫描8次,分辨率为2 cm-1。
1.2.8 计算机拟合分子包合 采用基于CHARMm的能量产生模型模拟CCN和HBCD的化学结构计算包合前后的能量变化及可能的构象[7]。
2 结果与讨论
2.1 含量测定方法建立 CCN储备液以乙醇稀释后的紫外吸收光谱见图1,由图1可知CCN在425 nm处有最大吸收峰。HBCD的乙醇液在425 nm处没有吸收,不干扰CCN测定,所以选择425 nm为测定波长。以CCN质量浓度(C)对吸光度(A)进行线性回归,得线性回归方程A=0.149 3C+0.005 8,R=0.999 9。 CCN在1.1~5.5 μg/mL浓度范围内与吸光度呈现良好的线性关系,见图2。日内精密度结果见表2,日间精密度结果见表3。从表2和表3中可以看出,溶液高、中、低浓度日内RSD为0.91%、0.35%、0.58%;日间RSD为1.23%、0.47%、0.75%,精密度符合要求。平均回收率分别为99.42%、100.14%、100.12%,RSD 为 0.45%、0.92%、1.17%,见表4。

图1 CCN的乙醇溶液紫外扫描图Fig.1 Ultraviolet scanning spectrum of CCN ethanolsolution
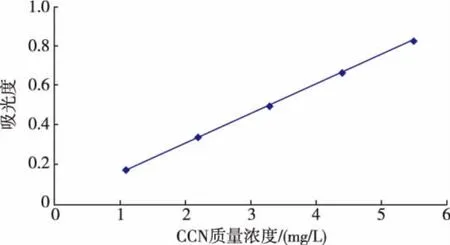
图2 CCN的无水乙醇溶液标准曲线Fig.2 Standard curve of the CCN in ethanol
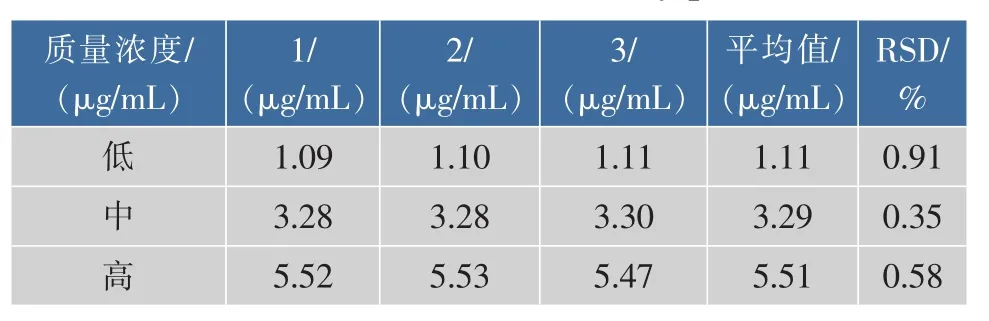
表2 日内精密度测定结果Table 2 Result of the intra-day precision
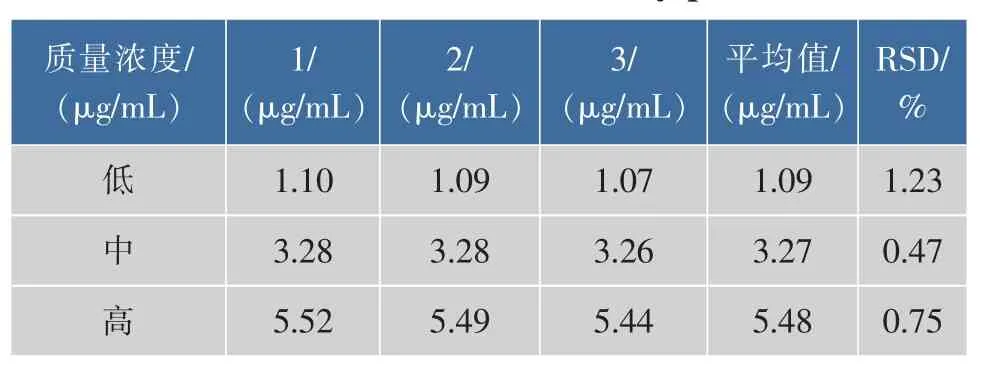
表3 日间精密度测定结果Table 3 Result of the inter-day precision

表4 回收率试验Table 4 Recovery experiment
2.2 CCIC最佳制备工艺的选择
按正交实验设计表进行试验,以CCN溶解度为考察指标,确定最佳制备工艺。正交实验结果见表5,方差分析结果见表6。
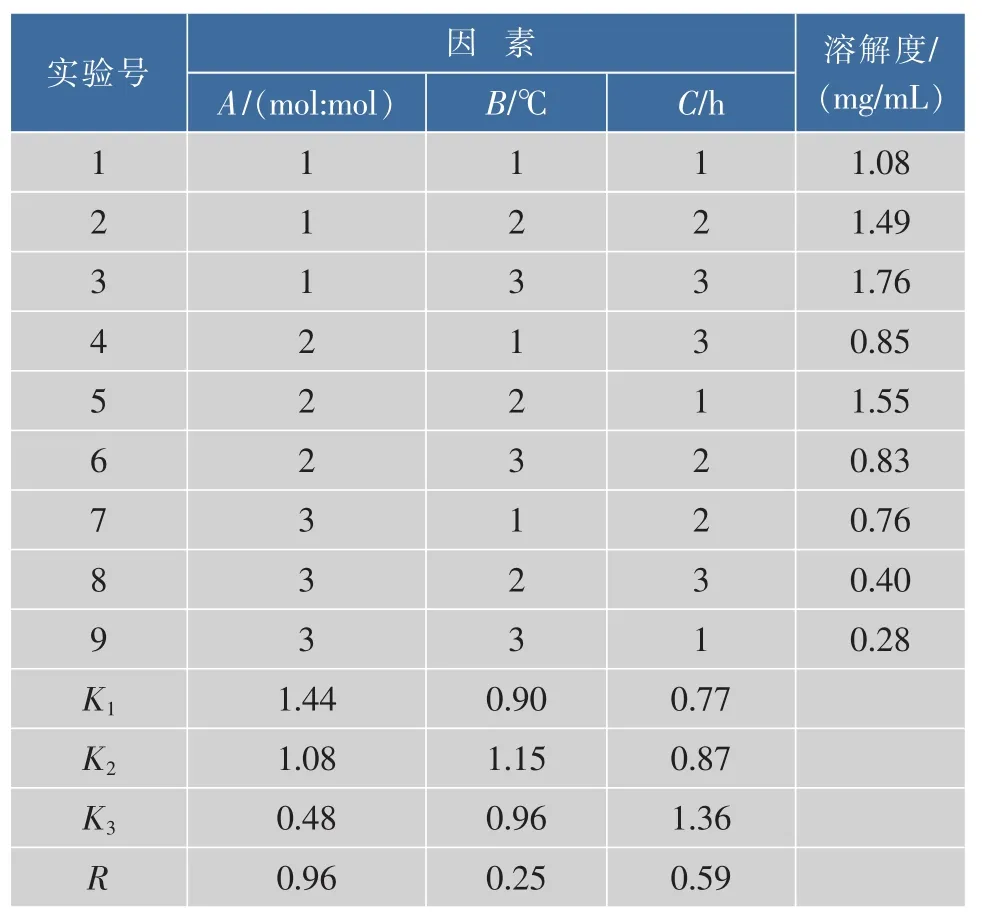
表5 正交试验结果Talle 5 Result of the results of orthogonal experiment
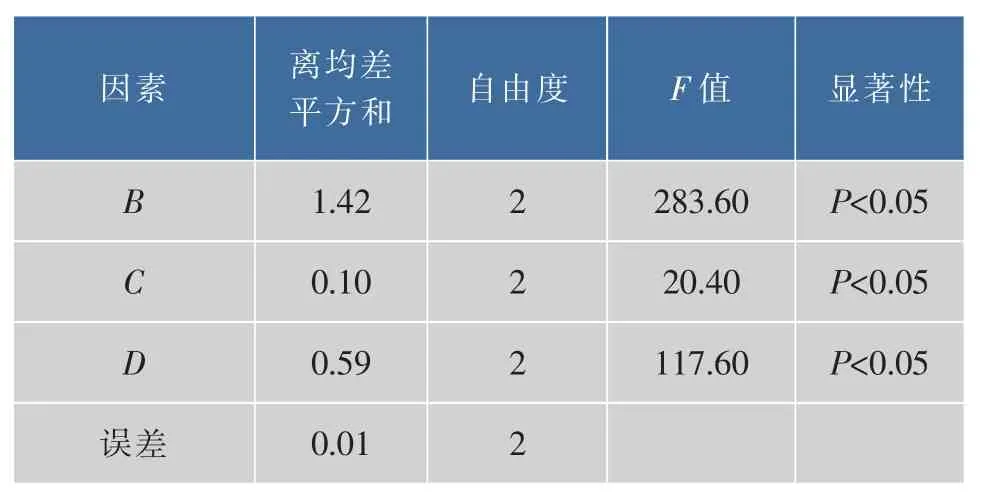
表6 方差分析结果Table 6 Results of analysis of variance
由表5正交实验结果可知因素B包合投料比的极差R最大,所以其对包合实验的影响最大,其次依次为因素D研磨时间和因素C包合时间。由表6方差分析结果可知3种因素对实验均有显著影响(P<0.05)。实验结果较大者为最优处方,根据结果确定最优处方工艺为B1C2D3。即包合投料摩尔比为1∶1,研磨温度为40℃,包合时间为1.5 h。按照最优化处方工艺B1C2D3重复制备3批CCIC,用“1.2.6溶解度测定”中方法测定每批CCIC溶解度,得到最优化处方工艺下CCN的溶解度为(1.72±0.03)mg/mL。
药物与环糊精一般形成1∶1或1∶2的包合物。由相溶解度法测定CCN与HBCD的包合常数,得到CCN与HBCD形成包合物的理论摩尔比为1∶1。而通过正交试验也验证了当CCN与HBCD的投料摩尔比为1∶1时,形成的CCIC有最大溶解度。
由正交试验得出制备CCIC的最优工艺为:包合投料摩尔比为1∶1,研磨温度为40℃,包合时间为1.5 h。随包合时间增加,包合效果也增加。而随温度的增加,包合效果却是先升高后降低,可能是由于CCIC在较高温度下不稳定。
2.3 相溶解度法测定CCN与HBCD的包合常数
相溶解度图见图3。
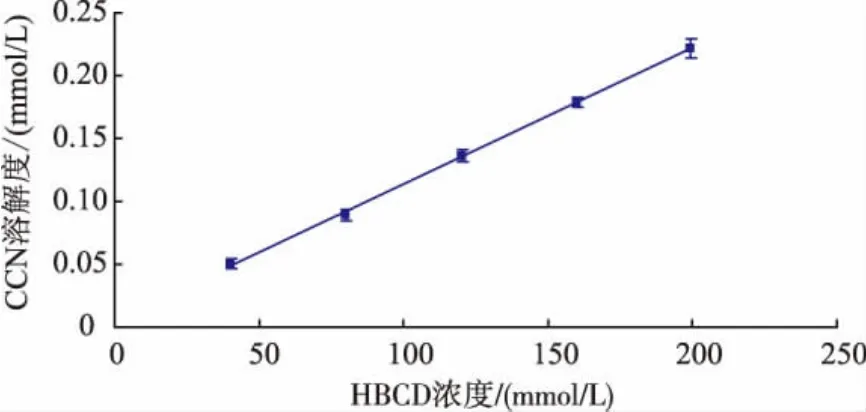
图3 CCN相溶解度曲线Fig.3 Phase solubility diagram of the CCN
由图3可知CCN溶解度随HBCD的增加而增加。CCN溶解度对HBCD浓度作图为直线,二者具有线性关系。直线方程为y=0.001x+0.000 4,R2=0.999 4>0.990 0为AL型。形成包和物的化学计量比为1∶1。由包合平衡常数K=斜率/截距(1-斜率)[8]得出 K=2.50×103L/moL。
K1∶1的数值范围在 0~1.00×105L/moL,包合平衡常数K越大,药物与环糊精形成的包合物越稳定。当包合平衡常数K小于80 L/moL时,则药物不适宜制备成包合物。相平衡溶解度法测得CCN与HBCD包合平衡常数K=2.50×103L/moL,包合平衡常数较大,形成的包合物较稳定。
2.4 溶解度测定
实验表明CCN达到溶解平衡的时间为30 min。按最优化处方工艺制备CCIC中CCN在蒸馏水中溶解度为(1.76±0.02) mg/mL(n=3)。
2.5 包合物的鉴定
显微观察法显示:在光学显微镜下观察,CCIC为不规则形状,明显不同于CCN和HBCD的简单物理混合物,证明形成了CCIC。差示量热扫描图谱见图4。CCN在175℃有一个吸热峰,为药物的熔融峰。CCN与HBCD物理混合物在该温度时有此吸热峰,而在CCIC中该吸热峰前移,峰型也明显减小,表明CCN与HBCD形成了包合物。红外分光光度法图谱见图5。通过比较药物包合前后在红外区吸收的特征差异,根据吸收峰的变化情况(吸收峰的降低、位移或消失),以此证明药物与环糊精是否产生包合作用。在包合物图谱中药物特征吸收峰1 207 cm-1峰消失,1 510、1 457、1 282、1 153cm-1峰强度明显减弱,表明CCIC形成。
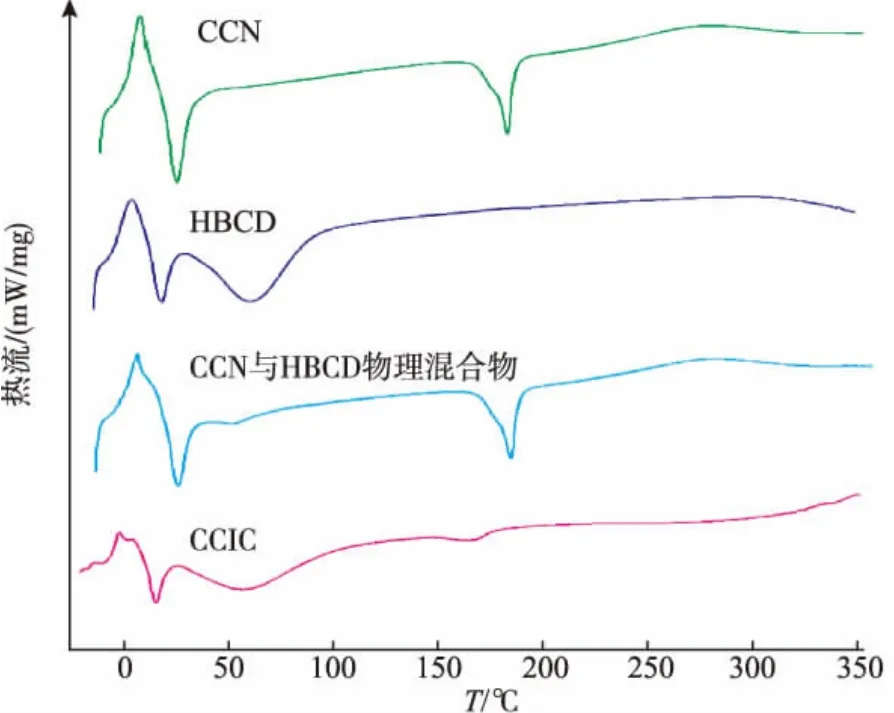
图4 差示扫描量热图Fig.4 Differential scanning colorimetry analysis
2.6 计算机拟合分子包合物
计算机模拟研究结果说明包合后体系能量下降了约175.728 kJ/mol,提示HBCD可能包合CCN后形成了CCIC,其可能的构象见图6。CCN和HBCD的化学结构包合前后的能量变化见表7。
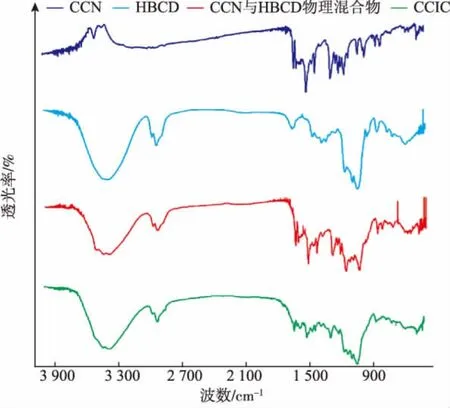
图5 红外吸收光谱图Fig.5 Infrared absorption spectrogram

图6 CCIC计算机拟合得到的构象图Fig.6 Computer fitting conformation
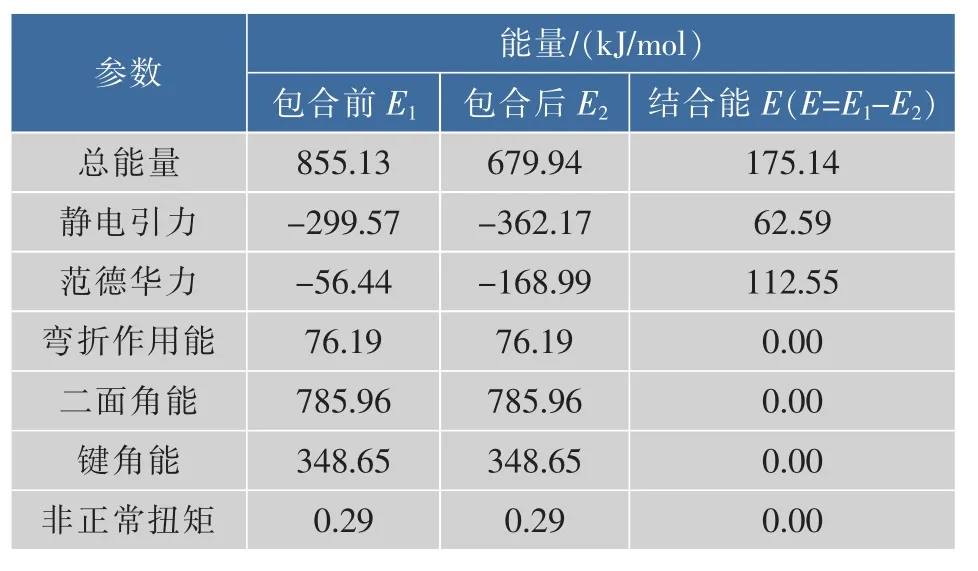
表7 分子拟合CCIC包合前后的能量变化Table 7 Energy changes of inclusive CCIC according to the molecular modeling
3 结 语
本实验通过预试验对饱和水溶液法、研磨法、超声法制备CCIC进行比较,得出研磨法制备的CCIC较其它两种方法制备的CCIC溶解度高,所以选用研磨法制备。HBCD水溶性较β-环糊精大[9],通过预试验对HBCD和β-环糊精进行了考察,研磨法制备HBCD和β-环糊精包合物,HBCD包合物溶解度明显高于β-环糊精包合物。所以试验选择以HBCD为材料研磨法制备CCIC。
CCN在蒸馏水中的溶解度为4.5×10-5mg/mL,制备成包合物后CCN溶解度增大了3.82×104倍。由此可见将CCN制备成CCIC,可大大提高CCN在水溶液中的溶解度。
[1]LIU Shan,TAN Qunyou,LIU Juan,JI Liu,et al.Study on taste-masking inclusion compound of dextromethorphan[J].Chin JMAP,2010,27(5):414-418.(in Chinese)
[2]PAULA D W X,DENADAIA M,SANTORO M M,etal.Supramolecularinteractionsbetweenlosartanand hydroxypropyl-β-CD:ESI mass-spectrometry,NMR techniques,phase solubility,isothermal titration calorimetry and anti-hypertensive studies[J].International Journal of Pharmaceutics,2011,404(1-2):116-123
[3]JIANG Y,SHA X,ZHANG W,et al.Complex of 9-nitro-camptothecin in hydroxypropyl-β-cyclodextrin:In vitro and in vivo evaluation[J].International Journal of Pharmaceutics,2010,397:116-121.
[4]QIAN L,GUAN Y,XIAO H.Preparation and characterization of inclusion complexes of a cationic β-cyclodextrin polymer with butylparaben or triclosan[J].International Journal of Pharmaceutics,2008,357:244-251.
[5]RANPISE N S,KULKARNI N S,MAIR P D,et al.Improvement of water solubility and in vitro dissolution rate of aceclofenac by complexation with beta-cyclodextrin and hydroxypropyl-beta-cyclodextrin[J].Pharmaceutical Development and Technology,2010,15:64-70.
[6]HIRLEKAR R,KADAM V.Preformulation study of the inclusion complex warfarin-β-cyclodextrin[J].International Journal of Pharmaceutics,2005,291:3-10.
[7]MATILAINEN L,TOROPAINEN T,VIHOLA H,et al.In vitro toxicity and permeation of cyclodextrins in Calu-3 cells[J].Journal of Controlled Release,2008,126:10-16.
[8]LIU Juan,TAN Qunyou,LU Weiping,et al.Study on inclusion process of rifampicin β-cyclodextrin inclusion complex[J].Chinese Journal of Hospital Pharmacy,2010,30:40-42.(in Chinese)
[9]ZHANG Li,TAN Qunyou,CHEN Liang,et al.Preparation of inclusion compound pyridostigmine bromide-β-cyclodextrin[J].Chinese Journal of Hospital Pharmacy,2010,30(6):466-470.(in Chinese)
