微波辅助衍生-气相色谱-质谱法同时测定养殖水体中6种雌激素
2018-01-18马丽莎李丽春谢文平刘书贵戴晓欣郑光明
马丽莎,李丽春,谢文平,单 奇,刘书贵,尹 怡,戴晓欣,郑光明
(中国水产科学研究院,珠江水产研究所/农业部热带亚热带水产种质资源利用与养殖重点实验室,农业部水产品质量安全风险评估实验室(广州), 广东 广州 510380)
雌激素是一类新型环境激素类污染物,普遍存在于我国自然水体中[1-3],其激素效应是其他常见内分泌干扰物的10 000~100 000倍[4],能促发多种癌症[5],对人体健康具有严重的潜在威胁。由于雌激素可通过水生生物富集,危害生态安全和人类健康,因此养殖水域的生态安全问题已成为社会关注的焦点。
目前,关于水环境中雌激素检测报道多集中于海水、江河及城市污水[6-8],尚未见养殖水体的报道。养殖水体的基质成分与上述水体差异较大,如藻类较多、水质粘性大、腐殖酸含量高等。雌酮(E1)、β-雌二醇(E2)、雌三醇(E3)是自然雌激素,炔雌醇甲醚(mestranol)、乙炔雌二醇(EE2)、己烯雌酚(DES)是人工合成的雌激素,它们都是口服避孕药、畜牧养殖用药的主要成分,也是水环境中安全隐患风险较高的雌激素。为加强我国水产养殖环境安全管理,建立上述6种雌激素在养殖水域中的检测方法势在必行。
雌激素的检测方法主要有高效液相色谱法(HPLC)、气相色谱-质谱法(GC/MS)及高效液相色谱-串联质谱法(HPLC-MS/MS)[9-14]等。HPLC法具有一定的普遍性,但其检出限较高; HPLC-MS/MS法与GC/MS法均具有较高的选择性和灵敏度,但液质很难排除基质干扰[15],且仪器价格昂贵;GC/MS法无基质效应且仪器价格适中,实验室普及率较高,但样品需衍生化,操作繁琐、耗时长,使其实际应用受到一定限制。近年来,通过分子极化和离子导电两个效应在短时间内完成样品加热的技术,在样品提取、消解、衍生等诸多领域得到了广泛应用[16-21]。
为改进GC/MS法衍生化的操作繁琐,本工作拟采用微波辅助技术与GC/MS法结合分析养殖水体中的6种雌激素残留,希望为养殖水体中痕量雌激素类物质的实时监测和批量分析提供技术支持。
1 实验部分
1.1 仪器与试剂
Agilent 7890A-5975C气相色谱-质谱联用仪:美国Agilent公司产品,配有电子电离源,HP-5MS柱(30 m×0.25 mm×0.25 μm);旋转蒸发仪:日本Eyela公司产品;GAST无油隔膜真空泵及12通道半自动固相萃取装置:美国嘉仕达公司产品;甲醇、乙腈、乙酸乙酯、二氯甲烷:均为色谱纯,美国Tedia公司产品;Supelclean ENVI-18 SPE小柱(500 mg/6 mL):德国Sigma-Aldrich公司产品;Oasis HLB 固相萃取柱(500 mg/6 mL):美国Waters公司产品;G70F 20CN1L-DG(S0)家用微波炉:广东格兰仕集团有限公司产品。
雌酮(E1)、雌三醇(E3)、炔雌醇甲醚(mestranol)、乙炔雌二醇(EE2)、β-雌二醇(E2)、己烯雌酚(DES)标准品:均为德国Dr.Ehrenstorfor公司产品;β-雌二醇-D2标准品(E2-D2):美国Sigma公司产品;己烯雌酚-D8标准品(DES-D8):加拿大Toronto公司产品。
衍生化试剂:N,O-双(三甲基硅烷基)三氟乙酰胺(BSTFA)/三甲基氯硅烷(TMCS)(99∶1,V/V),德国r-Biopham公司产品;N-甲基-N-三甲基硅基三氟乙酰胺(MSTFA):美国Regis公司产品。
1.2 标准储备液及标准工作液的配制
准确称取适量的雌激素标准品,分别用甲醇溶解并稀释,配制成浓度均为100 mg/L的单个标准储备液,于-20 ℃保存。准确移取适量的单个标准储备液,用甲醇稀释、定容,配制成6种激素浓度均为1 mg/L的混合标准工作液,于-20 ℃保存。
准确称取适量的内标,分别用甲醇溶解并稀释,配制成浓度均为100 mg/L的内标标准储备液,于-20 ℃保存。准确移取适量的单个内标标准储备液,用甲醇稀释、定容,配制成己烯雌酚-D8和雌二醇-D2浓度均为1 mg/L的混合内标工作液,于-20 ℃保存。
1.3 水样的采集与预处理
采集的水样保存于棕色瓶中,用HCl调至pH 4以避免微生物降解,保护水样,置于4 ℃冰箱中保存。
取5 000 mL实际水样,经0.45 μm纤维素膜过滤。取1 000 mL水样滤液,加入50 μL 1 mg/L的内标工作溶液,混合均匀。采用Supelclean ENVI-18 SPE固相萃取小柱,依次用12 mL甲醇、12 mL超纯水活化平衡后上样,控制过柱流速为3 mL/min。先用12 mL超纯水淋洗SPE小柱,再用10 mL正己烷淋洗SPE小柱,速度为3 mL/min,弃去淋洗液。真空干燥30 min后,用18 mL正己烷-二氯甲烷溶液(1∶1,V/V)以1 mL/min的流速分3次洗脱。收集洗脱液,于50 ℃下氮气吹干,向吹干的残留物中加入100 μL乙酸乙酯溶液,盖塞并涡旋混合20 s,再加入100 μL MSTFA衍生化试剂,盖塞并涡旋混合1 min,于700 W功率下微波衍生4 min,冷却至室温,待GC/MS分析。
1.4 实验条件
1.4.1色谱条件 色谱柱:HP-5MS柱(30 m×0.25 mm×0.25 μm);载气:高纯氦气,纯度大于99.999%,流速1.0 mL/min;进样口温度250 ℃;采用脉冲不分流进样,脉冲压力207 kPa,持续时间0.9 min,分流出口开启时间1 min,进样体积1 μL;升温程序:初始柱温50 ℃,保持1 min,以20 ℃/min升至220 ℃,保持27 min,以20 ℃/min升至250 ℃,保持20 min。
1.4.2质谱条件 离子源温度230 ℃,四极杆温度150 ℃,接口温度250 ℃,溶剂延迟10 min,选择离子监测模式(SIM)。
2 结果与讨论
2.1 质谱条件的选择
在m/z50~550范围内进行全扫描,确定各组分的保留时间和特征离子。为降低干扰、提高灵敏度,采用选择离子监测模式,每个化合物选择1个定量离子、3个定性离子。6种雌激素的保留时间、定性与定量离子及内标物列于表1。
2.2 质谱条件的优化
实验发现,采用普通进样方式分析样品中雌激素时,其重现性较差。这可能是由于雌激素为弱极性物质,其分子中的羟基易受进样系统的吸附而损失;且雌激素高温易分解,较高的进样口温度可能导致目标化合物的部分降解。而脉冲不分流进样方式通过瞬间提高进样口压力的方式,减少了样品在进样口的停留时间,降低了目标化合物被吸附和分解的风险,可确保进样量的精确度,以保证雌激素峰组分的重现性,提高灵敏度、改善峰形,更适于痕量、易分解化合物的残留分析。通过实验比较发现,脉冲不分流进样所得的目标化合物重现性、灵敏度均比普通不分流进样的好,峰形也更尖锐。因此,本实验选择脉冲不分流进样模式。
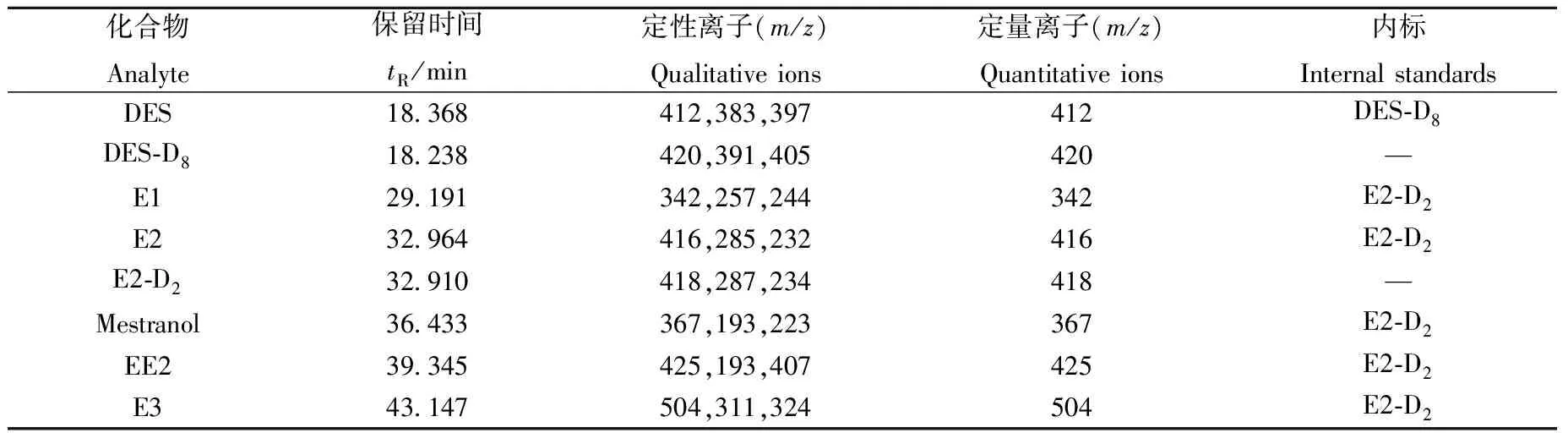
表1 6种雌激素的保留时间、定性离子、定量离子与内标物Table 1 Retention times, qualitative ions, quantitative ions and internal standards of 6 estrogens
选择合适的脉冲压力是获得目标峰最佳灵敏度、准确性及重现性的关键:脉冲压力过小对改善目标峰灵敏度、重现性的作用不大;过大会引起进样口隔垫过早损坏,导致进样口漏气,且隔垫碎渣掉入衬管易污染衬管,导致杂峰多,干扰目标物的测定,使重现性变差。实验发现,目标物灵敏度、重现性均随脉冲压力的增大而增大,但当脉冲压力大于207 kPa时,进样口隔垫受损过快。因此,经综合考虑,本实验选择脉冲压力为207 kPa。
2.3 SPE萃取条件的优化
2.3.1SPE小柱的选择 雌酮、雌三醇、炔雌醇甲醚、乙炔雌二醇、β-雌二醇、己烯雌酚均为疏水亲脂性化合物。Oasis HLB小柱由亲脂性的二乙烯苯和亲水性的N-乙烯基吡咯烷酮混合而成,可保留亲水和亲脂两方面的化合物,被广泛应用于饮用水、河流等自然水体中雌激素的残留分析[1-5]。但实验发现,HLB小柱应用于养殖水体中雌激素的残留分析时,其回收率不稳定,在50%~200%之间,相对标准偏差RSD>30%,且背景干扰大。鉴于雌激素是弱极性化合物,本实验选用Supelclean ENVI-18 SPE小柱萃取养殖水体中的雌激素,获得了较高的回收率88.5%~106%,相对标准偏差为0.29%~11.9%,且背景干扰小。分析原因,养殖水体受渔业活动影响较大,水体中的藻类、N、P等亲水性杂质含量普遍较高,而这些亲水性杂质可竞争HLB小柱上的活性位点,影响雌激素在小柱上的富集,导致其回收率不稳定。ENVI-18 SPE小柱主要通过目标物的碳氢键同硅胶表面的官能团产生非极性的范德华力来保留目标物,非常适用于水样中弱极性有机物的富集和纯化,其碳含量高达17%,可对弱极性化合物产生更大的键合能力,有利于增强小柱对化合物的吸附能力,提高小柱的回收率和稳定性。综上所述,本实验选择ENVI-18 SPE小柱富集样品中的雌激素组分。
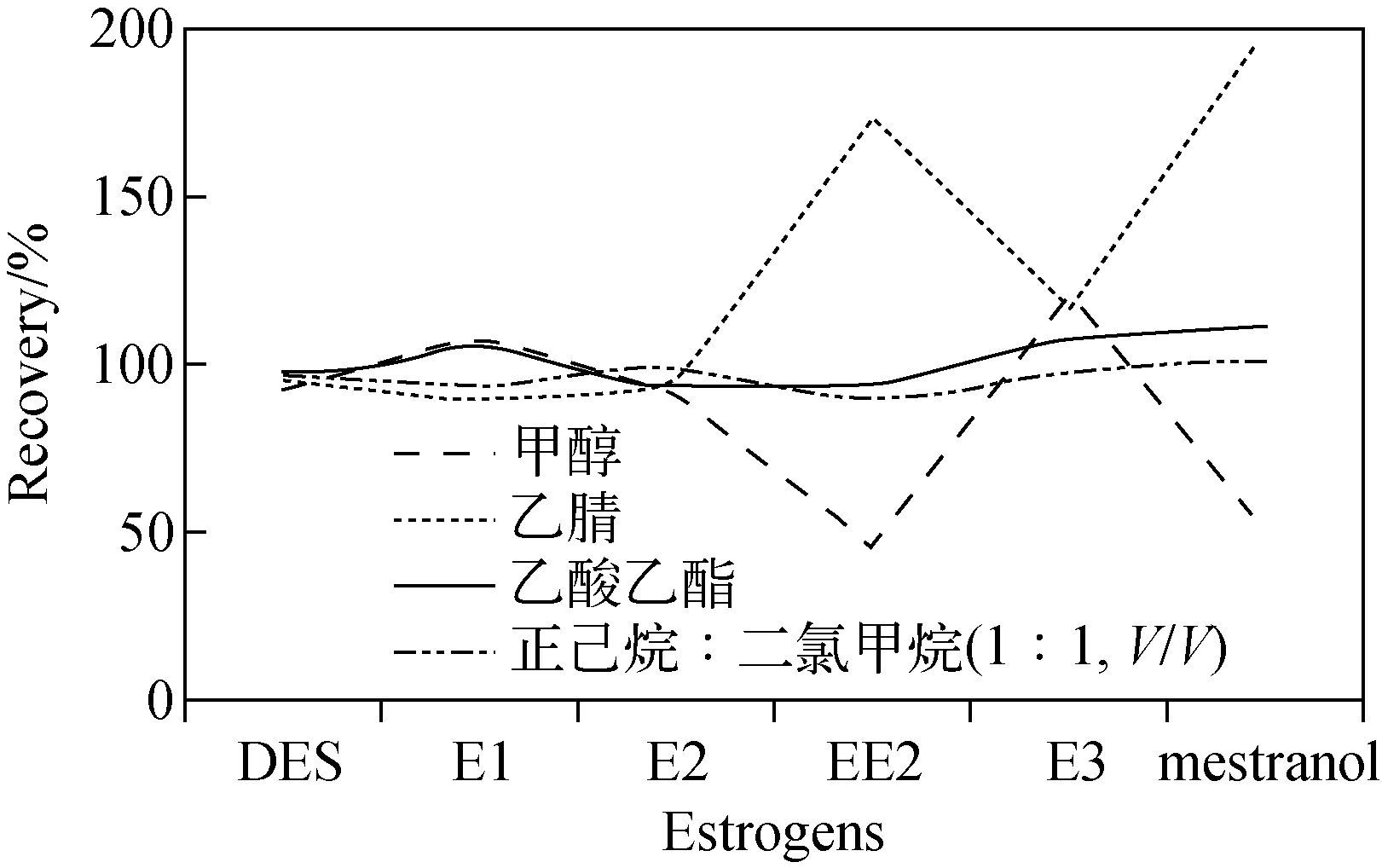
图1 不同洗脱溶剂对6种雌激素回收率的影响Fig.1 Effects of elution solvent on the recoveries of 6 estrogens
2.3.2洗脱剂和淋洗溶剂的选择 洗脱剂是决定回收率高低的关键因素。本实验比较了甲醇、乙腈、乙酸乙酯、正己烷-二氯甲烷溶液(1∶1,V/V)4种不同极性溶剂的洗脱效果,结果示于图1。使用甲醇时,EE2与 mestranol的回收率过低,仅有50%左右;而使用乙腈时,EE2与 mestranol的回收率又过高,达170%以上。因此,甲醇、乙腈均不适于作为雌激素的洗脱溶液。使用乙酸乙酯和正己烷-二氯甲烷溶液作为洗脱剂时,各雌激素的回收率均在100%左右,可满足实验要求,但乙酸乙酯的洗脱液颜色略呈黄色,图谱基线没有正己烷-二氯甲烷溶液的平整,杂峰也较多。因此,本实验选择正己烷-二氯甲烷溶液(1∶1,V/V)作为洗脱溶剂。
另外,由于养殖水样基质复杂,有必要在洗脱之前对ENVI-18 SPE小柱进行淋洗。本实验先用12 mL超纯水淋洗SPE小柱,除去富集在小柱上的亲水性杂质,再用10 mL脂溶性强的正己烷淋洗SPE小柱,除去小柱上色素等非极性杂质,最后用正己烷-二氯甲烷(1∶1,V/V)溶液洗脱,从而获得比较理想的色谱图。
2.4 衍生条件的优化
2.4.1衍生化方法的确定 传统的雌激素衍生方法是烘箱加热或水浴加热,均是由外部热源通过热辐射由表及里进行传导式加热,衍生反应时间长、衍生效率低且能耗高,极大地限制了GC/MS法的应用。微波辅助衍生技术通过分子极化和离子导电两个效应对物质直接加热,消除了一般加热过程中由于电热板、空气、容器壁的热传导和热辐射造成的热量损失,具有较高的传热效率。本实验采用该技术加速雌激素的衍生化反应,与传统加热法的衍生效果对比图示于图2。可见,微波辅助衍生法所得6种雌激素衍生物的峰面积均大于烘箱加热法,增强了GC/MS法检测雌激素的灵敏度,微波衍生法的衍生化效率比烘箱加热法的提高了50%左右,雌激素的衍生化时间从烘箱加热的90 min缩短到4 min。
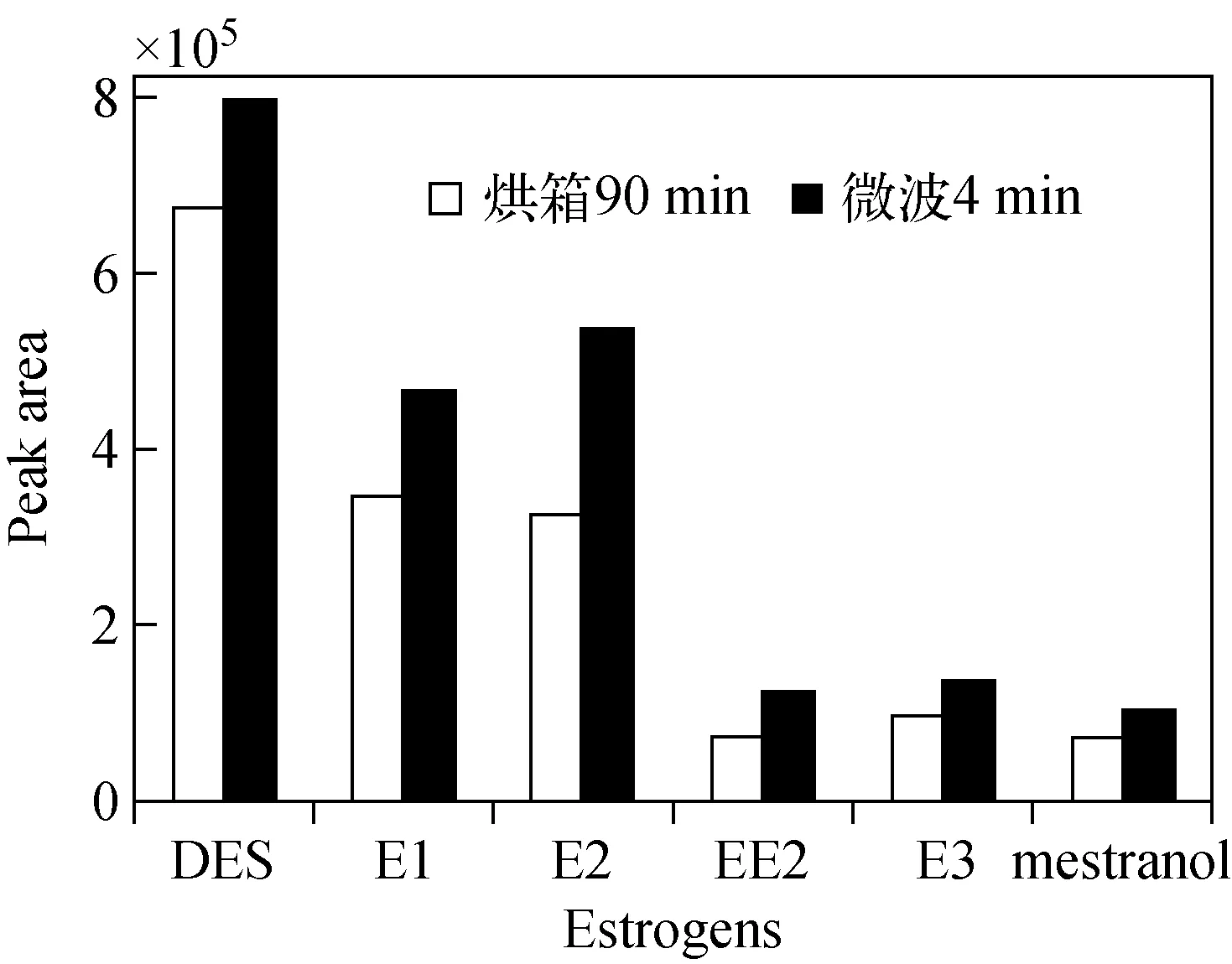
图2 不同衍生方式对衍生效果的影响Fig.2 Effects of different derivatize methods
2.4.2衍生化试剂的选择 分别选用MSTFA与BSTFA-TMCS(99∶1,V/V)两种常用的硅烷化试剂与雌激素分子的羟基进行硅烷化反应。结果表明,MSTFA可与6种雌激素发生较好的衍生化反应,而BSTFA-TMCS虽能与E1、E2、E3、DES的羟基进行硅烷化反应,但不与mestranol、EE2发生反应。根据化合物的结构进行分析,推测其原因为mestranol、EE2分子的脂族羟基碳链上连有其他基团,BSTFA分子较大,不能与mestranol、EE2的反应基团有效结合,而MSTFA分子较小,不受空间位阻效应的影响,能与之结合,得到硅烷化产物。因此,本实验选用MSTFA为衍生化试剂。此外,极性溶剂能更好地吸收微波,可增强衍生反应效率。本实验考察了乙腈、乙酸乙酯和正己烷对雌激素衍生效率的影响,结果表明,当乙酸乙酯作为衍生溶剂时,雌激素的衍生效率最高且稳定性更好。因此,选用乙酸乙酯作为雌激素的衍生溶剂。
2.4.3微波衍生化方法的优化 微波衍生技术是利用微波加热的特性对样品中的目标化合物进行衍生反应。该方法优化的关键因素是衍生化反应时间:反应时间过短,对待测物的衍生反应不彻底;反应时间过长,衍生产物在微波辐照下会发生降解,且基体中的许多杂质被衍生而生成更多的副产物,产生大量干扰。本实验在微波700 W时,比较了衍生化时间分别为2、3、4、5 min时,MSTFA的衍生化效率,结果示于图3。可见,随着衍生化反应时间的增加,雌激素衍生化效率逐步提高,在4 min时达最大值,之后开始下降。为防止目标化合物降解,保障其最佳的衍生化效率,本实验选择的衍生化条件为700 W衍生4 min。
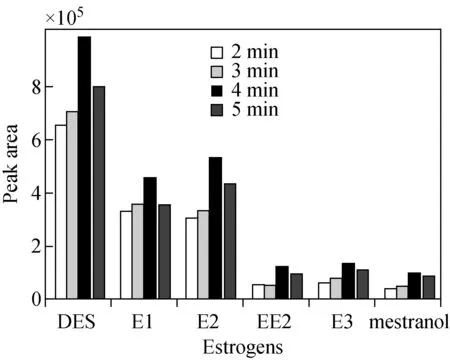
图3 不同衍生时间对衍生效果的影响Fig.3 Effects of different derivatize times
2.5 线性关系、方法的检出限
配制质量浓度分别为1、2、5、25、50、125、250、500 μg/L的8个水平的混合标准工作溶液,取100 μL标准溶液,加入50 μL混合内标使用液,氮气吹干,之后分别加入100 μL乙酸乙酯溶液及100 μL MSTFA衍生化试剂进行衍生化。在选定的色谱条件和质谱参数下分析,以目标物的定量离子峰面积和相应内标物的定量离子峰面积之比(y)为纵坐标,以目标物浓度(x,μg/L)为横坐标,进行线性回归分析,以信噪比(S/N)为3,结合回收率计算方法检出限。结果表明,雌激素DES在2~500 μg/L,E2在1~500 μg/L,E1、E3、EE2、mestranol在5~500 μg/L范围内均呈现良好的线性关系,相关系数(R2)均大于0.999 3。6 种化合物的线性范围、线性方程、相关系数和检出限列于表2。
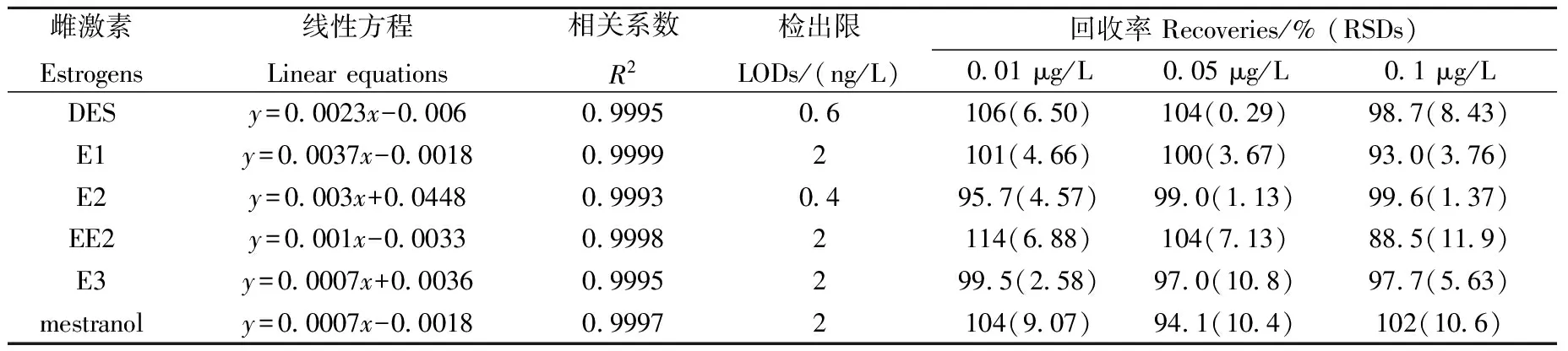
表2 6种雌激素的线性回归方程、相关系数、检出限及3个添加水平的回收率和相对标准偏差(n=6)Table 2 Linear equations, correation coefficients (R2), limits of detection (LODs), recoveries and relative standard deviations (RSDs) of 6 estrogens in water for aquaculture spiked at three leves (n=6)
2.6 回收率与精密度
取1 L经0.45 μm滤膜过滤的养殖用水,用上述方法测出目标化合物含量,作为空白对照组。然后对同一水样进行3个不同浓度的加标实验,加标量分别为0.01、0.05、0.1 μg/L,测定目标化合物含量。扣除空白含量计算方法回收率,结果列于表2。6种雌激素的平均回收率为88.5%~106%,相对标准偏差为0.29%~11.9%,均小于15%。因此,所建立的方法具有较好的准确度和精密度,可满足分析要求,所得的加标回收色谱图示于图4。
2.7 实际样品的检测
将本实验建立的方法用于20份池塘养殖水体中雌激素残留检测,在其中3份池塘养殖水体中检出己烯雌酚残留,含量分别为0.65、0.80和2.8 ng/L,其余雌激素组分无检出。己烯雌酚作为人造求偶素,在水产养殖中常用作生长促进剂。 由此可见,渔业行为对养殖水体雌激素含量有影响,养殖水体环境中的雌激素可能由渔业生产者因盲目追求高产量低成本,不恰当使用求偶素、排卵素等药物而引起。
3 结论
建立了微波辅助衍生-气相色谱-质谱法检测养殖水体中6种痕量雌激素,该方法利用微波加热的特性衍生样品中的目标化合物,改进了传统GC/MS法在样品前处理中衍生化操作繁琐、耗时长的不足,使衍生化时间从烘箱加热的90 min缩短到4 min,提高了样品前处理的效率,具有高效、快速、灵敏度高、特异性强、节能环保的优点,可满足养殖水体中痕量雌激素类物质实时监测的需要,适用于养殖水体中雌激素类化合物的批量分析。
[1] 吴世闽,贾瑗,彭辉,等. 辽东湾海水中甾体雌激素的检测及生态风险评价[J]. 中国环境科学,2011,31(11):1 904-1 909.
WU Shimin, JIA Ai, PENG Hui, et al. Determination and risk assessment of steroidal estrogens in Liaodong Bay, China[J]. China Environmental Science, 2011, 31(11): 1 904-1 909(in Chinese).
[2] 宋文婷,陆光华,李湘鸣,等. 长江(南京段)环境雌激素的污染特征[J]. 生态环境学报,2009,18(5):1 615-1 619.
SONG Wenting, LU Guanghua, LI Xiangming, et al. Characters of environmental estrogen pollution in Yangtze River (Nanjing section)[J]. Ecology and Environmental Sciences, 2009, 18(5): 1 615-1 619(in Chinese).
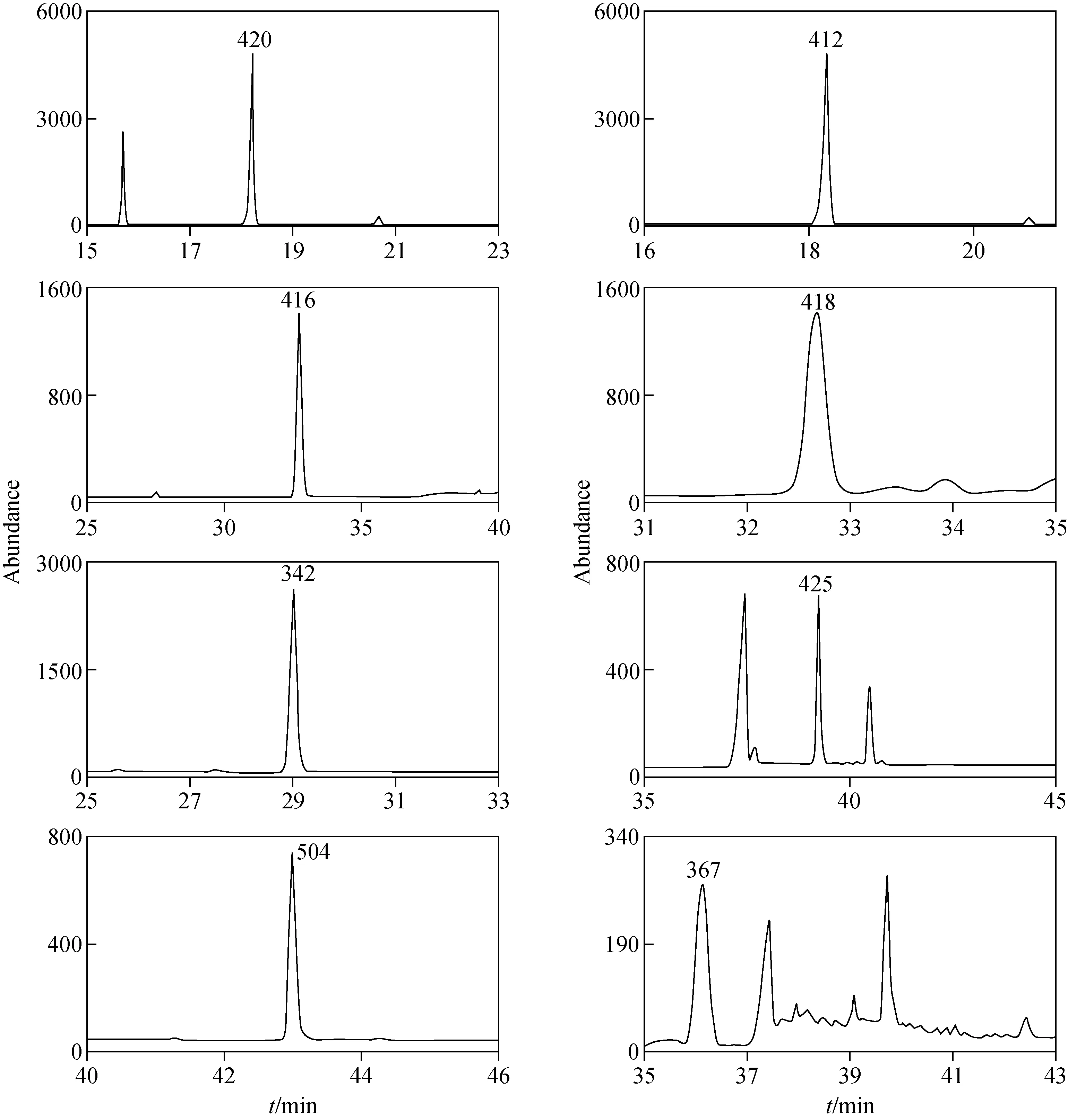
图4 选择离子监测模式下,养殖用水的加标回收色谱图Fig.4 Selective ion monitor (SIM) chromatograms of water for aquaculture spiked with estrogens
[3] 田怀军,舒为群,邱志群,等. 长江流域某市饮用水雌激素污染物初步研究[J]. 第三军医大学学报,2004,26(19):1 751-1 754.
TIAN Huaijun, SHU Weiqun, QIU Zhiqun, et al. Primary study of estrogenic pollutants in drinking water in a city on the Yangtze River[J]. Acta Academiae Medicinae Militaris Tertiae, 2004, 26(19): 1 751-1 754(in Chinese).
[4] HANSELMAN T A, GRAETZ D A,WILKIE A C. Manure-borne estrogens as potential environmental contaminants: a review[J]. Environmental Science Technology, 2003, 37(24): 5 471-5 478.
[5] FEIGELSON H S, HENDERSON B E. Estrogens and breast cancer[J]. Careinogenesis, 1996, 17(11): 2 279-2 284.
[6] QUINTANA J B, CARPINTEIRO J, RODR′IGUEZ I, et al. Determination of natural and synthetic estrogens in water by gas chromatography with mass spectrometric detection[J]. Journal of Chromatography A, 2004, 1 024(1/2): 177-185.
[7] LIU S, YING G G, ZHAO J L, et al. Trace analysis of 28 steroids in surface water, waste water and sludge samples by rapid resolution liquid chromatography-electrospray ionization tandem mass spectrometry[J]. Journal of Chromatography A, 2011, 1 218(10): 1 367-1 378.
[8] CHEN B B, HUANG Y L, HE M, et al. Hollow fiber liquid-liquid-liquid microextraction combined with high performance liquid chromatography-ultraviolet detection for the determination of various environmental estrogens in environmentaland biological samples[J]. Journal of Chromatography A, 2013, 1 305(1): 17-26.
[9] 廖涛,吴晓翠,王少华,等. 固相萃取-气相色谱/质谱联用法同时检测水体中9种环境雌激素[J]. 分析化学研究简报,2013,41(3):422-426.
LIAO Tao, WU Xiaocui, WANG Shaohua, et al. Simultaneous detection of nine kinds of estrogens in water by solid phase extraction coupled with gas chromatography-mass spectrometry[J]. Chinese Journal of Analytical Chemistry, 2013, 41(3): 422-426(in Chinese).
[10] 谭丽超,葛峰,单正军,等. 超高效液相色谱-串联质谱法测定污水处理厂水样中的雌激素[J]. 生态与农村环境学报,2011,27(5):86-92.
TAN Lichao, GE Feng, SHAN Zhengjun, et al. Determination of estrogens in water samples from wastewater treatment plant using ultra-performance liquid chromatography-electrospray tandem mass spectrometry method[J]. Journal of Ecology and Rural Environment, 2011, 27(5): 86-92(in Chinese).
[11] KUMIRSKA J, PLENIS ALINA,UKASZEWICZ P, et al. Chemometric optimization of derivatization reactions prior to gas chromatography-mass spectrometry analysis[J]. Journal of Chromatography A, 2013, 1 296(12): 164-178.
[12] RAO K F, LEI B L, LI N, et al. Determination of estrogens and estrogenic activities in water from three rivers in Tianjin, China[J]. Journal of Environmental Sciences, 2013, 25(6): 1 164-1 171.
[13] 赵云芝,钱蜀,谢永洪,等. 固相萃取-超高效液相色谱-串联质谱法测定水中6种雌激素[J]. 中国环境监测,2013,29(1):107-111.
ZHAO Yunzhi, QIAN Shu, XIE Yonghong, et al. Determination of six estrogens in water using solid phase extraction-ultra high performance liquid chromatography-tandem mass spectrometry[J]. Environmental Monitoring in China, 2013, 29(1): 107-111(in Chinese).
[14] 那广水,陈彤,张月梅,等. SPE-HPLC-MS/MS法检测水体和沉积物中六种甾体雌激素[J]. 海洋环境科学,2011,30(3):424-427.
NA Guangshui, CHEN Tong, ZHANG Yuemei, et al. Determination of six estrogens in water and sediment by SPE-HPLC-MS/MS[J]. Marine Environmental Science, 2011, 30(3): 424-427(in Chinese).
[15] YAN W, ZHAO L X,FENG Q Z, et al. Simultaneous determination of ten estrogens and their metabolites in waters by imroved two-step SPE followed by LC-MS[J]. Chromatographia, 2009, (69): 621-628.
[16] 郭双双,杨利民,张一鸣,等. 微波辅助萃取人参总皂苷与单体皂苷含量分析[J]. 食品科学,2015,36(2):1-5.
GUO Shuangshuang, YANG Limin, ZHANG Yiming, et al. Determination of contents of total saponins and monomer saponins inPanaxginsengby microwave-assisted extractiion method[J]. Food Science, 2015, 36(2): 1-5(in Chinese).
[17] 赖莺,林睿,蔡鹭欣,等. 微波辅助萃取-气相色谱-质谱法测定丙烯酸树脂中9种残余单体[J]. 色谱,2012,36(1):21-26.
LAI Ying, LIN Rui, CAI Luxin, et al. Determination of 9 residual acrylic monomers in acrylic resins by gas chromatography-mass spectrometry coupled with microwave assisted extraction[J]. Chinese Journal of Chromatography, 2012, 36(1): 21-26(in Chinese).
[18] 李攻科,何小青,张展霞. 微波消解同时衍生化GC-MS法测定血浆中脂肪酸的研究[J]. 分析测试学报,2000,19(1):16-18.
LI Gongke, HE Xiaoqing, ZHANG Zhanxia. Determination of fatty acids in plasma sample by GC-MS using microwave-assisted digestion and derivatization[J]. Journal of Instrumental Analysis, 2000, 19(1): 16-18(in Chinese).
[19] 林维宣,董伟峰,陈溪,等. 气相色谱-质谱法同时检测动物组织中多种激素类兽药的残留量[J]. 色谱,2009,27(3):294-298.
LIN Weixiong, DONG Weifeng, CHEN Xi, et al. Determination of hormonemulti-residues in animal tissues by gas chromatography-massspectrometry[J]. Chinese Journal of Chromatography, 2009, 27(3): 294-298(in Chinese).
[20] ZUO Y, ZHANG K, LIN Y. Microwave-accelerated derivatization for the simultaneous gas chromatographic-mass spectrometric analysis of natural and synthetic estrogenic steroids[J]. Journal of Chromatography A, 2007, 1 148(2): 211-218.
[21] MOMENBEIK F, KHORASANI J H. Separation and determination of sugara by reversed-phase high-performance liquid chromatography after pre-column microwave-assisted derivatization[J]. Analytical and Bioanalytical Chemistry, 2006, 384(3): 844-850.
