亲水性Obelisc R液相色谱-串联质谱法测定猪肉样品中氨基糖苷类药物残留
2018-01-18赵凤娟方恩华韩瑞阳彭毅候熊贝贝蔡伊娜李丽苏
赵凤娟,方恩华,韩瑞阳,彭毅候,罗 耀,熊贝贝,蔡伊娜,李丽苏
(1.深圳出入境检验检疫局食品检验检疫技术中心, 深圳市食品安全检测技术研发重点实验室,广东 深圳 518045;2.厦门出入境检验检疫局检验检疫技术中心,福建 厦门 361026)
氨基糖苷类抗生素是由氨基糖与氨基环醇通过氧桥连接而形成的一种苷类抗生素药物[1],临床上主要用于治疗敏感需氧革兰阴性杆菌所致的全身感染。由于氨基糖苷类药物存在确切的耳毒性和肾毒性,近年来在人类抗感染临床上的应用已越来越少。随着我国畜牧业的发展和动物源性食品产量的增加,氨基糖苷类抗生素作为廉价的广谱抗生素,被广泛应用于动物疫病防治。但该类药物具有良好抗菌作用的同时,也具有严重的不良反应。其毒副作用主要表现为肾毒性、耳毒性、神经肌肉毒性和过敏反应等诸多不良反应,其中最为严重的是耳毒性。上世纪六七十年代,因氨基糖苷类抗生素广泛使用,导致大批病人失聪。为了保障食品安全,目前,我国农业部、美国FDA、欧盟委员会及日本厚生劳动省对常用的几种氨基糖苷类抗生素的残留限量标准做出了规定,限定其在猪肉中的最大残留量在50~600 μg/kg之间。中国作为全球动物源性食品生产和进出口大国,建立一套先进、高效、准确的检测方法势在必行。
氨基糖苷类抗生素的化学结构中存在多个氨基和羟基基团,具有强极性和弱碱性,其水溶性较好,能与无机酸或有机酸生成盐,以硫酸盐或盐酸盐的形式存在[2]。目前对于动物源性食品中氨基糖苷类抗生素残留检测的主要方法有酶联免疫吸附法(ELISA)、 微生物法、气相色谱法、液相色谱法、高效液相色谱法、高效液相色谱-质谱联用法、毛细管电泳法等。王忠斌等[3]以新霉素的降解产物新霉胺作为半抗原,采用戊二醛法合成新霉胺免疫原,制备出针对多种氨基糖苷类药物的多克隆抗体;并使用高碘酸盐法制备新霉胺的酶标抗原,建立了氨基糖苷类药物的直接竞争酶联免疫方法。Verheijen等[4]建立了竞争ELISA测定链霉素,利用胶体金标记纯化的链霉素单克隆抗体 (mAb),竞争抗原为牛血清白蛋白和链霉素的连接物,链霉素的最低检测限为60 μg/L。这些方法前处理简单、快捷,适用于动物源性食品中氨基糖苷类抗生素残留的快速检测,但不能进行准确定量和确证。Hoebus等[5]建立了气相色谱法测定壮观霉素盐酸盐的含量,由于前处理衍生化过程复杂,方法回收率不高,该方法现已很少使用。由于氨基糖苷类抗生素的特殊理化性质,在紫外区段没有特征,需经衍生化后,用反相色谱或离子对色谱系统进行分离检测。陈晓红等[6]用NaOH和1,2-蔡醒-4-磺酸钠溶液双试剂柱后衍生,以荧光检测器在激发波长263 nm和发射波长447 nm处进行测定;蔡玉娥等[7]用离子交换柱分离妥布霉素、新霉素和西索霉素,并用脉冲积分安培电化学检测器进行检测;苑丽等[8]建立了高效液相色谱法结合电化学检测器测定动物源食品中大观霉素的残留量。由于氨基糖苷类抗生素化学结构中含有多个伯胺或仲胺基团,易在离子源中被电离成正离子,近年来多使用高灵敏度的高效液相色谱-串联质谱法进行检测,但氨基糖苷类药物的极性大,在一般的反相色谱柱上基本无保留,需要添加七氟丁酸等增强其保留[9]。Bogiallid等[10-13]分别建立了牛奶、肉、内脏等食品中氨基糖苷类药物的液相色谱-串联质谱测定方法,均采用向流动相中添加七氟丁酸的方式增加氨基糖苷类药物的保留。离子对试剂的长期使用会严重抑制质谱仪的电喷雾离子化效果,大幅降低仪器灵敏度,尤其是对负离子模式的影响更为明显。为了解决此问题,近年来有多篇文献报道采用亲水性色谱柱(如HILIC等)分离氨基糖苷类药物,例如,Turnipseed等[14]通过将氨基糖苷类药物经异氰酸苯酯衍生化的方式,建立了牛奶中氨基糖苷类药物的液相色谱-离子阱质谱测定方法;Ishii等[15]采用ZIC-HILIC液相色谱柱分离,串联质谱测定肾脏和肉中6种氨基糖苷类药物;Kawano[16]采用亲水性HILIC色谱柱建立了样品中链霉素和双氢链霉素的液相色谱-电喷雾三重四极杆飞行时间质谱分析方法。
本工作拟采用一种新型的亲水性Obelisc R色谱柱,选择乙腈-水溶液作为流动相,通过调节乙腈与酸的比例对氨基糖苷类药物进行分离,结合高专属性的氨基糖苷分子印迹固相萃取柱前处理净化,建立猪肉中壮观霉素(spectinomycin, SPE)、潮霉素B(hygromycin B, HYG)、双氢链霉素(dihydrostrepmycin, DHSTR)、链霉素(streptomycin, STR)、阿米卡星(amkacin, AMI)、卡那霉素(kanamycin, KAN)、安普霉素(apramycin, APR)、妥布霉素(tobramycin, TOB)和庆大霉素(gentamicin, GEN)9种氨基糖苷类药物的高效液相色谱-串联质谱测定方法。
1 实验部分
1.1 主要仪器与装置
Nexera®UHPLC LC-30A 高效液相色谱仪:日本岛津公司产品;6500 Qtrap串联质谱仪:美国AB Sciex公司产品,配有ESI电喷雾离子源及AnalystTM1.6.2仪器工作站;固相萃取装置:美国Agilent公司产品;电子天平:瑞士Mettler Toledo公司产品;移液器:规格10~200 μL、100~1 000 μL、1~5 mL,德国Eppendorf公司产品;Biotage TurboVap®氮吹浓缩仪:美国Caliper公司产品;涡旋振荡器:美国Barnstead公司产品;高速冷冻离心机:德国Sigma公司产品;Milli-Q®实验室超纯水制备系统:美国Millipore公司产品。
1.2 材料与试剂
Obelisc R®高效液相色谱柱(2.1 mm×150 mm×5 μm,100 Å):美国Sielc公司产品;SupelMIP®SPE固相萃取柱:50 mg/3 mL,美国Sigma-Aldrich公司产品;0.22 μm有机微孔滤膜:中国津腾公司产品;乙腈、甲醇、甲酸:均为色谱纯,德国Merck公司产品;乙酸铵、三氯乙酸:均为色谱纯,德国Sigma-Aldrich公司产品;磷酸二氢钠、乙二胺四乙酸二钠、浓氨水:均为分析纯,天津科密欧化学试剂有限公司产品。
安普霉素标准品(纯度81.5%),硫酸庆大霉素标准品(纯度94.4%),链霉素标准品(纯度96%),硫酸卡那霉素标准品(纯度98%),双氢链霉素标准品(纯度99.0%),阿米卡星标准品(纯度100%),壮观霉素标准品(纯度99%),潮霉素B标准品(纯度70%):均为德国Dr. Ehrenstorfer GmbH公司产品;妥布霉素标准品(纯度97.2%):日本TCI公司产品。
标准储备液的制备:分别准确称取适量的每种氨基糖苷类标准品,用水溶解,配成浓度为100 mg/L 的标准储备溶液,于4 ℃避光可保存6个月。
混合标准中间溶液的制备:分别准确量取1 mL各标准储备液于10 mL容量瓶中,用水定容至刻度,混匀,于4 ℃避光可保存1个月。
样品提取液的制备:准确称取1.36 g磷酸二氢钾,用980 mL水溶解,并用1.0 mol/L的盐酸调pH至4.0,然后分别加入0.15 g Na2EDTA和20 g三氯乙酸,溶解混匀并定容至1 000 mL;Aminoglycosides-SPE样品洗脱液的制备:准确量取800 mL乙腈,加入200 mL水并充分混合,加入10 mL甲酸和660 μL七氟丁酸,超声溶解混匀。
1.3 实验条件
1.3.1色谱条件 色谱柱:SiELC Obelisc R®亲水性高效液相色谱柱(2.1 mm×150 mm×5 μm,100 Å);流动相:A为1%甲酸水溶液, B为90%乙腈-水溶液;流速400 μL/min;柱温40 ℃;进样量20 μL;梯度洗脱程序:0~0.5 min(5%A),0.5~5 min(A线性增至45%,保持1 min),5~8.5 min(A线性增至95%,保持2.5 min),8.5~8.6 min(A线性降至5%,保持3.4 min)。
1.3.2质谱条件 电喷雾离子源(ESI),正离子扫描,多反应监测模式;离子源喷雾电压5 500 eV;离子源温度450.0 ℃;气帘气(N2)压力206.8 kPa;雾化气(N2)压力344.7 kPa;辅助加热气(N2)压力413.7 kPa;锥孔电压10.0 eV;碰撞室出口电压9.0 eV;氨基糖苷类药物定性、定量离子,碰撞能量等其他参数列于表1。
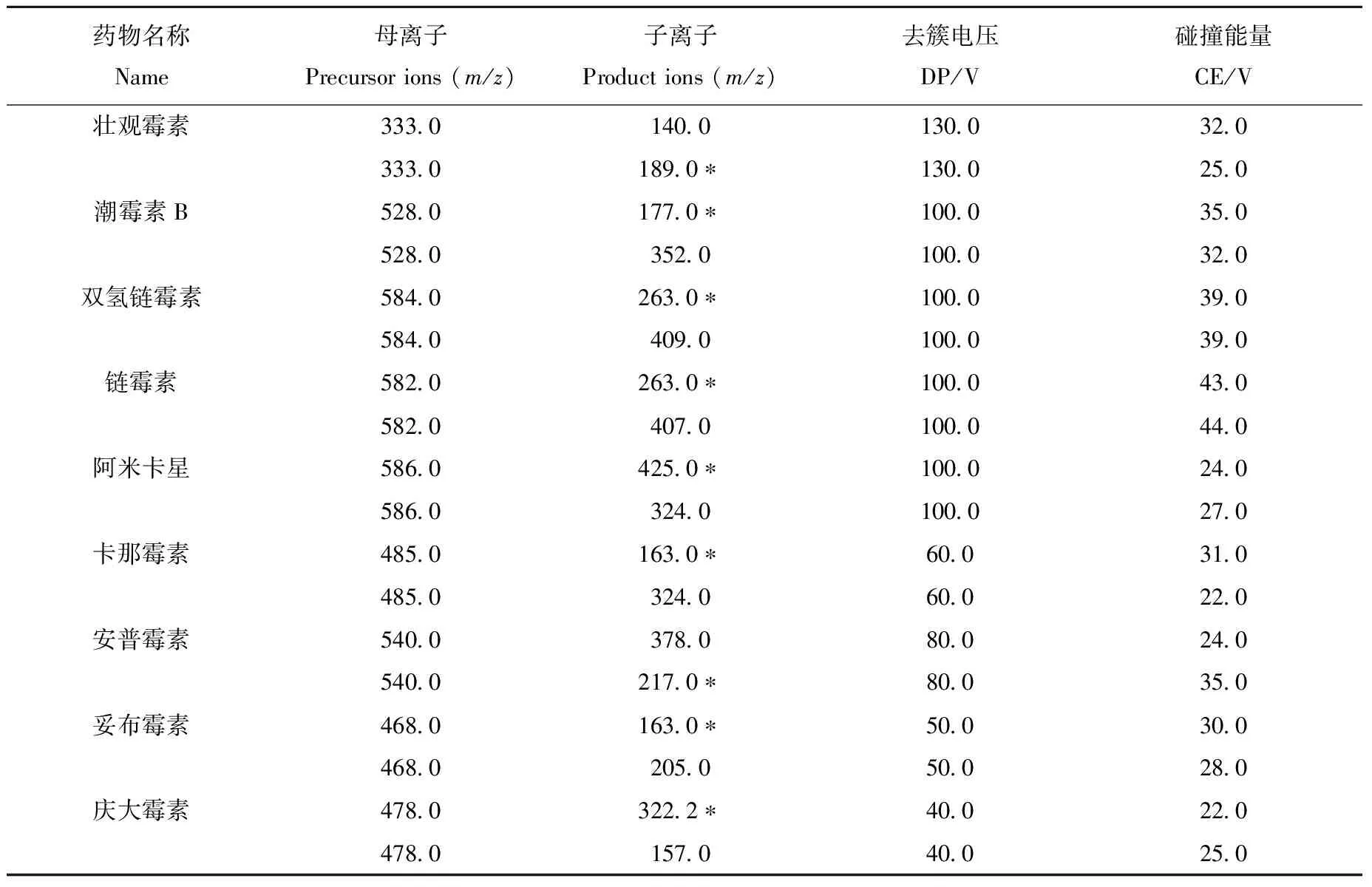
表1 氨基糖苷类药物的定性、定量离子及去簇电压、碰撞能量Table 1 ESI settings for HPLC-MS/MS analysis of the AGs
注:*为定量离子
1.4 样品前处理方法
1.4.1提取 称取2 g(精确到0.01 g)试样置于50 mL聚丙烯离心管中,加入10.0 mL样品提取液涡旋振荡2 min,并于平板振荡器上振荡提取10 min,以9 000 r/min离心5 min,精确吸取5.0 mL上清液并转移至另一个50 mL聚丙烯离心管中,用稀氨水调pH至7.5±0.2,涡旋混匀。
1.4.2净化 SupelMIP®SPE-Aminoglycosides固相萃取柱先后用1 mL甲醇、1 mL 50 mmol/L磷酸盐缓冲液(pH 7.8)淋洗活化,将1.4.1节中调节pH值后的提取液加载在固相萃取柱上,控制流速约1滴/秒,先用3 mL水淋洗后真空抽干2~3 min,然后用1 mL乙腈-水溶液(4∶6,V/V)淋洗,弃去淋洗液并抽干10 s,最后用1 mL二氯甲烷-甲醇溶液(1∶1,V/V)淋洗并抽干10 s。用2 mL Aminoglycosides-SPE样品洗脱液洗脱,收集洗脱液于精密刻度离心管中,于45 ℃水浴下氮气吹干部分溶剂,用乙腈定容至1.0 mL,涡旋混匀后,过0.22 μm有机微孔滤膜,待测。
1.4.3基质匹配工作曲线 取空白猪肉样品,按1.4.1节的方法进行提取,经SPE净化后,用空白基质液稀释混合标准溶液,配制出0、20、50、100、200、500、1 000 μg/L 9种浓度的氨基糖苷混合标准工作液。以标准工作液的浓度为横坐标,各分析物定量子离子的峰面积为纵坐标,绘制标准曲线。
2 结果与讨论
2.1 质谱条件的优化
由于氨基糖苷类抗生素的化学结构中含有多个伯胺或仲胺基团,具有弱碱性,易在质谱仪离子源中被电离成正离子,同时,由于该类药物含有多个羟基基团,具有很强的极性,常采用电喷雾(ESI)离子源进行分析。本实验采用ESI离子源,在正离子扫描模式下,采用流动注射的方式分别对9种氨基糖苷类药物进行母离子扫描(Q1 scan),结合各药物的分子式及扫描质谱图确定各药物的准分子离子;然后分别以其准分子离子为母离子,对其子离子进行全扫描(Product Ion Scan),选取丰度较强、干扰较小的子离子为定性离子,每种化合物至少选择两对离子,以满足欧盟96/23/EC指令中对残留检测确证的要求。在多反应监测模式下,对各离子对的去簇电压、碰撞能量以及离子源参数等进行优化,确定最佳质谱条件(于1.3.2节中列出)。
2.2 液相色谱条件的优化
氨基糖苷类药物极性很强,在反相C18色谱柱中的保留行为很弱,需要在流动相中加入三氟乙酸、七氟丁酸等离子对试剂增强保留并改善峰形,但离子对试剂的使用会造成质谱端极强的离子抑制(尤其对于负离子),对离子源污染十分严重,同时会导致色谱柱柱效及寿命的下降、色谱峰重现性较差等问题。为了防止离子对试剂对质谱仪电喷雾离子化的抑制作用,需要寻找其他分离原理的色谱柱。随着近年来亲水性(如HILIC等)色谱柱的发展和应用,分离极性化合物的方法更加多样化。本实验采用一种新型亲水性Obelisc R色谱柱,在反相模式下分离氨基糖苷类药物。Obelisc R色谱柱是基于液态分离池技术(LiSCTM)的一种新型色谱柱,填料为特殊细孔结构硅胶,细孔内部有较大表面积,通过人工化学修饰方法结合其上带电基团,形成一个带有正电荷、具有较强离子强度和疏水特性的液态分离池。类似于活细胞与外界环境存在平衡,液态分离池存在于一个与流动相动态平衡的环境中,分析物通过硅胶孔进出分离池而达到保留目的,分离池特性随着外部(流动相)的化学环境改变而改变,包括产生长疏水链和亲水链、离子化程度改变、电荷分布和反离子性质的改变等。其丰富的离子交换特性使得分离方法的开发具有多样性,仅通过改变乙腈-水的比例或酸浓度即可实现对分析物保留行为的改变,甚至改变洗脱顺序。
在亲水性色谱分离模式下,一般釆取富含有机相的流动相,无论固定相是否被电离,只要流动相中水的比例大于1%就可以在固定相表面出现多层吸附,形成“富水层”,该“富水层”的厚度满足极性样品在流动相和固定相中的分配。在有机溶剂中,乙腈作为流动相,可以获得较好的样品保留及尖锐对称的峰形,是多数方法的首选。由于流动相中乙腈-水的比例对Obelisc R色谱柱的影响较大,因此重点探究了流动相中不同比例有机相对分离效果的影响。水相采用1%甲酸溶液。考察流动相中乙腈比例分别为50%、70%、90%时,500 μg/L 氨基糖苷混合标准溶液的保留情况,结果示于图1。
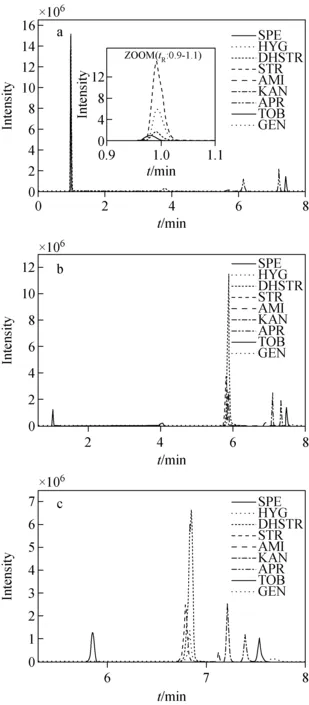
图1 流动相中含50%乙腈(a)、70%乙腈(b)、90%乙腈(c)的混合标准溶液色谱图(500 μg/L)Fig.1 Chromatograms of 50%ACN (a), 70%ACN (b) and 90%ACN (c) in mobile phase
由图1可以看出,乙腈比例为90%时各物质均有较好的保留,分离效果较好。随着乙腈比例的降低,各物质的保留时间减小,70%乙腈时,壮观霉素保留较弱;50%乙腈时壮观霉素、潮霉素B、链霉素、双氢链霉素均无保留。庆大霉素随着乙腈比例的降低,峰形明显变差,信号变弱。因此本实验采用乙腈-水溶液(9∶1,V/V)作为流动相。
除乙腈的比例外,水相中酸的浓度对分离也有显著影响。在流动相中添加不同浓度的甲酸溶液,在优化的洗脱条件下,考察Obelisc R对氨基糖苷类抗生素的分离效果,结果示于图2。
由图2可知,甲酸浓度的增大可以增强流动相的洗脱能力;随着酸浓度的降低,庆大霉素、妥布霉素、阿米卡星的响应明显减弱,峰形展宽,不利于定量测定。综合考虑9种物质的保留情况、响应强度、峰形、色谱柱耐受程度等因素,选择添加1.0%的甲酸为最佳条件。
2.3 基质效应的评价
喷雾电离离子源(ESI)容易受样品基质的影响,在分析生物样品时,未消除基质效应的影响可能会造成检测结果偏高或偏低,所得的实验结果不可靠。基质效应存在基质增强或基质减弱。
本研究采用提取添加法对基质效应进行评价,将氨基糖苷类标准溶液分别用空白溶剂(A1)与未净化的空白猪肉样品提取液(A2)稀释至200 μg/L,然后进行HPLC-MS/MS测定,考察基质效应,结果示于图3。
未经净化的猪肉样品提取液对氨基糖苷类抗生素质谱信号影响非常大,而且由结果可知, SPE、HYG、DHSTR、STR四种物质出现明显的基质信号抑制效应,其中抑制效应最严重的为STR,抑制比值仅为0.36;而AMI、KAN、APR、TOB、GEN五种物质有明显的基质信号增强效应,APR增强比值高达3.75。
2.4 样品前处理条件的优化
为减少基质效应的影响,对样品提取液进行有效净化十分必要。目前文献报道对氨基糖苷类药物的净化多采用C18固相萃取小柱,而分子印迹聚合物固相萃取吸附剂(molecularly imprinted polymer sorbents)是近年来发展的一种将固相萃取与分子印迹技术结合起来的一种材料,对特定的化合物有较强的选择性。本研究采用SupelMIPTMSPE-Aminoglycosides固相萃取小柱对样品进行净化,并与C18固相萃取净化的效果进行了比较。分别用溶剂稀释标液(STD)、不经固相萃取净化的猪肉样品提取液(Without SPE)、经C18固相萃取小柱净化的样品提取液(C18)以及经SupelMIPTMSPE-Aminoglycosides固相萃取小柱净化提取液(SupelMIPTMSPE)稀释的200 μg/L标准溶液进行HPLC-MS/MS分析。通过比较信号强度考察净化效果,结果示于图4。
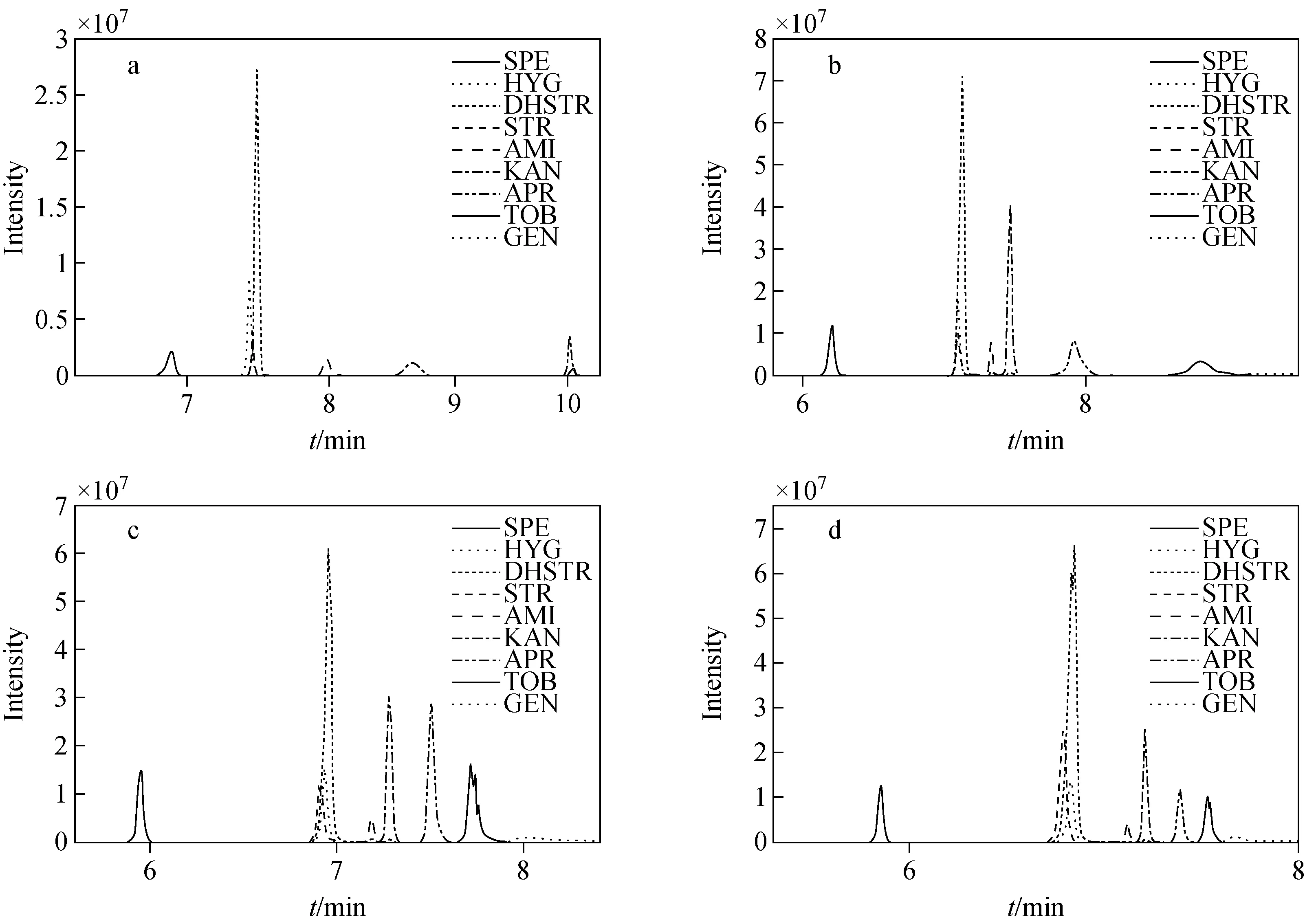
图2 流动相中含0.4%甲酸(a)、0.6%甲酸(b)、0.8%甲酸(c)和1.0%甲酸(d)的混合标准溶液色谱图Fig.2 Chromatograms of 0.4%FA (a), 0.6%FA (b), 0.8%FA (c) and 1.0%FA (d) in mobile phase

图3 猪肉基质对药物响应信号的影响Fig.3 Pork matrix effect on response signals

图4 不同净化方式对分析物响应的影响Fig.4 Purification method effect on response signals
由图4可知,对于存在基质抑制的4种药物(SPE、HYG、DHSTR和STR)经过净化以后,基质抑制情况得到改善,采用分子印迹固相萃取法改善效果最为明显;而对于存在基质增强的5种药物(AMI、KAN、APR、TOB和GEN),经过净化后,基质增强效应均减弱,氨基糖苷分子印迹固相萃取净化法较经典C18净化效果更佳,这是由于其对分析物的高度选择性,可在淋洗步骤中尽可能多地去除干扰物,净化效果可提高10%~50%。
2.5 线性范围与定量限
在使用外标法进行定量时,为了尽可能使标准溶液与样品溶液具有相同的离子化效率,消除基质效应带来的测定值误差,本方法采用空白样品提取液作为标准溶液的配制溶剂。精密量取一定体积的浓度为10 mg/L的混合标准中间溶液,用处理后的空白基质提取液稀释,配制成一系列浓度分别为20、50、100、200、500和1 000 μg/L的标准溶液。混匀后,按已优化的色谱、质谱条件分别进样20 μL进行分析,所有分析物的质量浓度x(μg/L),和峰面积y之间均有良好的线性关系,相关系数(R2)在0.993 7~0.999 9之间。具体的标准曲线参数列于表2。
根据我国法规要求,猪肉中氨基糖苷类药物的残留最高限量在100~2 000 μg/kg之间,在空白猪肉样品中添加一定浓度的标准溶液,经前处理后上机测定,参考各种药物的残留限量标准,同时满足信噪比(S/N)大于10的要求,确定各化合物的定量限为50.0 μg/kg。
2.6 方法回收率与精密度
对猪肉样品分别进行3个浓度水平(50、200、500 μg/kg)的空白加标回收率实验,每个浓度水平进行6次平行测试,根据各组分的峰面积,计算相应浓度的回收率和精密度,测得数据列于表3。样品的平均添加回收率在76.9%~89.4%之间,精密度在3.56%~11.4%之间,可以满足分析要求。
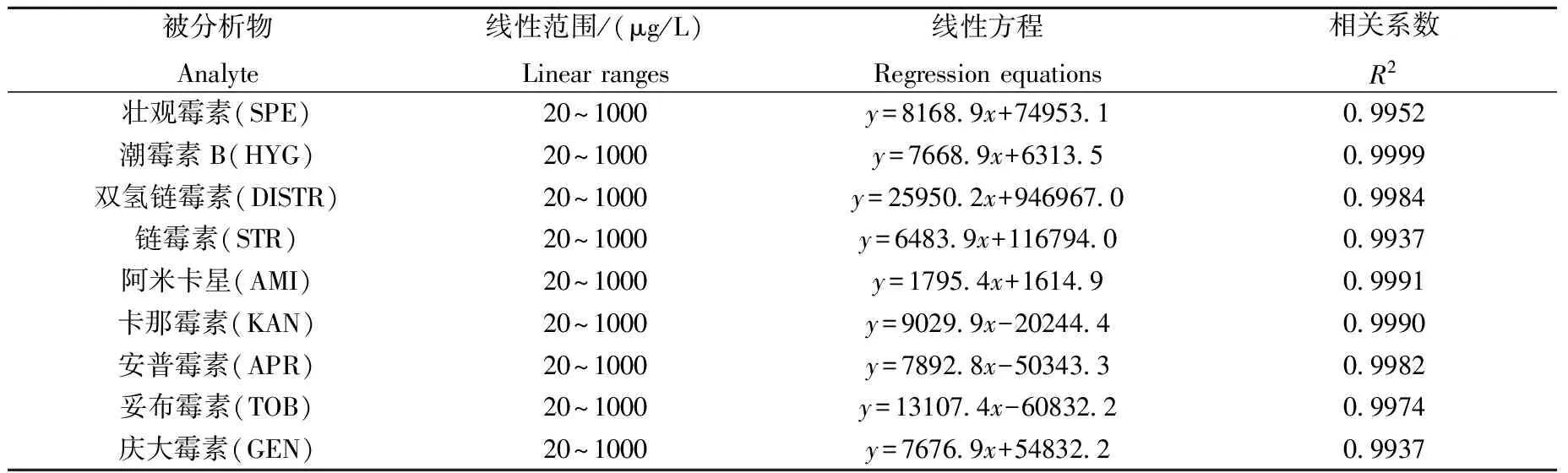
表2 线性方程和线性范围Table 2 Regression equations and linear ranges of 9 AGs
3 结论
采用了一种新型的亲水性Obelisc R色谱柱,以乙腈-水体系作为流动相,通过新型液态分离池模式下形成的富水层,使9种氨基糖苷类药物得到了很好的保留和分离,并用高灵敏度的串联质谱进行检测,避免了离子对试剂的引入。同时在前处理中引用了分子印迹固相萃取技术,提高了前处理的净化效率,降低了基质对氨基糖苷类药物检测的影响。在20~1 000 μg/L范围内线性关系良好,样品的回收率在76.9%~89.4%之间,相对标准偏差在3.56%~11.4%之间。该方法简便、快速、准确、灵敏度高,可以满足猪肉中氨基糖苷类药物残留的测定需求。
[1] 金有豫. 药理学[M]. 第5版. 北京:人民卫生出版社,2001:985-987.
[2] FORGE A, SCHACHT J. Aminoglycoside antibiotics[J]. Audiology and Neurotology, 2000, 5(1): 3-22.
[3] 王忠斌,王向红,徐蓓,等. 氨基糖苷类药物多残留酶联免疫分析方法的研究[J]. 中国食品学报,2008,8(5):120-125.
WANG Zhongbin, WANG Xianghong, XU Bei, et al. Studies on the ELISA for the simultaneous detection of aminoglycoside antibiotics[J]. Journal of Chinese Institute of Food Science and Technology, 2008, 8(5): 120-125(in Chinese).
[4] VERHEIJEN R, OSSWALD I K, DIETRIE R, et al. Development of a one step strip test for the detection of (dihydro)streptomycin residues in raw milk[J]. Food and Agriculture Immu nology, 2000, 12(1): 31-40.
[5] HOEBUS J, YUN L M, HOOGMARTENS J. An improved gas chromatographic assay for spectinomycin hydrochloride[J]. Chromatographia, 1994, 39(5): 71-73.
[6] 陈晓红,刘小莉,董明盛,等. 高效液相色谱法测定蜂产品中链霉素残留量[J]. 食品科学,2004,25(1):155-158.
CHEN Xiaohong,LIU Xiaoli,DONG Mingsheng,et al. New method on streptomycin residues assay in honey by HPLC[J]. Food Science, 2004, 25(1): 155-158(in Chinese).
[7] 蔡玉娥,蔡亚岐,牟世芬,等. 离子交换-脉冲积分安培法分离检测氨基糖苷类抗生素[J]. 分析实验室,2006,25(8):7-9.
CAI Yu’e,CAI Yaqi,MOU Shifen,et al. Separation of several aminoglycoside antibiotics with ion-exchange column following with pulsed integrated amperometric detection[J]. Chinese Journal of Analysis Laboratory, 2006, 25(8): 7-9(in Chinese).
[8] 苑丽,胡功政,肖传斌,等. 反相高效液相色谱测定动物源食品中大观霉素残留[J]. 中国抗生杂志,2007,32(3):166-168.
YUAN Li, HU Gongzheng, XIAO Chuanbin, et al. Determination of spectinomycin residues in animal food by RP-HPLC[J]. Chinese Journal of Antibiotics, 2007, 32(3): 166-168 in Chinese.
[9] GB/T 21323—2007 动物组织中氨基糖苷类药物残留量的测定高效液相色谱-质谱/质谱法[S]. 北京:中国标准出版社,2007.
[10] HOLTHOON F L V, ESSERS M L, MULDER P J, et al. A generic method for the quantitative analysis of aminoglyeosides (and spectinomycin) in animal tissue using methylated intemal standards and liquid chromatography tandem mass spectrometry[J]. Analytica Chimica Acta, 2009, 637: 135-143.
[11] BOGIALLI S, CURINI R, DI C A, et al. Simple confirmatory assay for analyzing residues of aminoglycoside antibiotics in bovine milk: hot water extraction followed by liquid chromatography-tandem mass spectrometry[J]. Journal of Chromatography A, 2005, 1 067(1/2): 93-100.
[12] KAUFMANN A, MADEN K. Determination of 11 aminoglycosides in meat and liver by liquid chromatography with tandem mass spectrometry[J]. Journal of Aoac International, 2005, 88(4): 1 118-1 125.
[13] KAUFMANN A, BUTCHER P, MADEN K. Determination of aminoglycoside residues by liquid chromatography and tandem mass spectrometry in a variety of matrices[J]. Analytica Chimica Acta, 2012, 711(711): 46-53.
[14] TURNIPSEED S B, CLARK S B, KARBIWNYK C M, et al. Analysis of aminoglycoside residues in bovine milk by liquid chromatography electrospray ion trap mass spectrometry after derivatization with phenyl socyanate[J]. Journal of Chromatography B Analytical Technology, 2009, 877(14/15): 1 487-1 493.
[15] ISHII R, HORIE M, CHAN W, et al. Multi-residue quantitation of aminoglycoside antibiotics in kidney and meat by liquid chromatography with tandem mass spectrometry[J]. Food Additives and Contaminants, 2008, 25(12): 1 509-1 519.
[16] KAWANO S. Analysis of impurities in streptomycin and dihydrostreptomycin by hydrophilic interaction chromatography/electrospray ionization quadrupole ion trap/time-of-flight mass spectrometry[J]. Rapid Communications in Mass Spectrometry, 2009, 23(6): 907-914.
