吡唑锌配合物的合成、晶体结构及抗肝癌细胞活性
2018-01-04吴春阳申贵男金成浩
李 婧 吴春阳 张 莹 申贵男 金成浩
(黑龙江八一农垦大学生命科学技术学院,大庆 163319)
吡唑锌配合物的合成、晶体结构及抗肝癌细胞活性
李 婧*吴春阳 张 莹 申贵男 金成浩
(黑龙江八一农垦大学生命科学技术学院,大庆 163319)
通过溶剂挥发法合成了锌配合物 [Zn(Hppo)2(ppo)Cl](1),Hppo=5-苯基-3-羟基吡唑,利用元素分析、FTIR以及X射线衍射法对配合物1进行表征。晶体结构显示配合物属于单斜晶系,P21空间群。检测了配合物1对肝癌细胞HepG2、Hep3B和Huh7的细胞形态与细胞毒性的影响,结果表明配合物1对肿瘤细胞具有明显的抑制作用。
吡唑;抗肿瘤活性;金属配合物;晶体结构
0 引 言
吡唑啉酮化合物具有多种生物活性,例如消炎、抗菌、抗肿瘤等,在医药、农药等方面得到广泛的应用[1-6],原因在于其具有多种配位形式,可以与多种金属形成结构各异的配合物。研究发现吡唑类金属配合物常表现出较吡唑配体更强的生物活性[6-7],其抗肿瘤作用使其成为抗肿瘤新药开发领域的热点[3-13]。例如Caruso等[10]报道了吡唑啉酮-5氧钛配合物,对乳腺癌细胞TA-3有较强的抑制作用,IC50为 90 μmol·L-1。 Budzisz等[11]合成 Pt和 Pd 的吡唑配合物,对人早幼粒白血病HL-60细胞和淋巴白血病NALM-6细胞均有较强的抑制作用,IC50均小于10 μmol·L-1。稀土金属在与吡唑啉酮形成配合物后也表现出抗肿瘤活性,何其庄等[12]报道的Gd配合物对人白血病K562细胞有明显的体外增值抑制作用(IC50=7.5 μmol·L-1)。吡唑啉酮金属配合物的开发和其抗肿瘤活性作用研究具有重要价值[14]。
锌是生物体内必需的微量元素,参与多种新陈代谢。在对非铂类抗癌药物的研究中,锌配合物的研究还较少[14-19]。 Zhang等[18]以 1-(3-(2-吡啶基)吡唑-1-基甲基)萘为配体合成吡唑类锌配合物,该配合物能有效的结合DNA,并且具有抑制癌细胞增值的作用。仇晓阳等[19]报道肼基二硫代甲酸苄酯席夫碱的锌配合物对肿瘤细胞MKN45有弱抑制性。Kovala-Demertzi等[20]合成了一组锌与胺苯硫脲类配合物,表现出具有和顺铂相当的抗肿瘤活性。Stanojkovic等[21]合成的4-氟苯基哌嗪锌的配合物对肿瘤增值效果强于顺铂,IC50值在 26~90 nmol·L-1。 Lopes等[22]以吡啶基缩氨基硫脲化合物为配体与锌形成配合物,体内抗肿瘤研究表明,配合物对MDA-MB-231和HepG2细胞具有较强的肿瘤抑制作用。锌类配合物的抗肿瘤活性研究具有重要的研究意义。
5-苯基-3-羟基吡唑(Hppo)结构当中含有2个N原子,一个氧原子,具有多种配位形式,其合成简单。以此化合物为配体,形成金属配合物并研究磁学性质早有报道[23],但未见抗肿瘤活性研究。本文以Hppo为配体与ZnCl2反应,得到单核配合物[Zn(Hppo)2(ppo)Cl](1),并初步用体外实验研究配合物对人肝癌细胞HepG2、Hep3B和Huh7的抑制作用,金属配合物表现出明显的肿瘤抑制活性。
1 实验部分
1.1 实验仪器、试剂与药品
元素分析在Perkin-Elmer 240C元素分析仪上测定。晶体结构在Bruker Smart APEX CCDⅡ衍射仪上完成。红外光谱使用Nicolet 380型红外光谱仪由KBr压片法测定。酶标仪使用Bio-Tek公司酶标仪。
试剂和药品:DMEM高糖(美国Hyclone公司);胎牛血清(Solarbio公司);胰蛋白酶(Solarbio公司);青链霉素(Solarbio公司);5-氟尿嘧啶(Macklin公司);台盼蓝(Sigma 公司);PBS 粉末(Biosharp 公司);MTT(二甲基溴化四氮唑蓝,Macklin公司);人肝癌细胞(HepG2,Hep3B,Huh7)取自美国模式培养物集存库(ATCC)。其他试剂均为分析纯。配体Hppo由水合肼和苯乙酰乙酸乙酯通过文献方法制备[23]。
1.2 配合物[Zn(Hppo)2(ppo)Cl](1)的合成
在圆底烧瓶中加入无水ZnCl2(0.068 g,0.5 mmol),无水乙醇(6 mL),在64℃下搅拌直至全部溶解。随后加入5-苯基-3-羟基吡唑(Hppo)[24](0.080 g,0.55 mmol),搅拌回流1 h,过滤,滤液静置于无震动的平台,用保鲜膜将烧杯封口,并在保鲜膜上扎若干小孔,1周后有白色块状晶体产物,产量0.028 g,产率26%。元素分析按C27H23ClN6O3Zn计算,实测值(%,括号内为计算值)C 55.23(55.88),H 4.10(3.99),N 14.32(14.48)。IR(KBr,cm-1):3 422m,3 339m,3 145m,1 618m,1 567m,1 535s,1 501s,756s,696m,548m。
1.3 晶体结构测定
取适宜大小的配合物1的单晶,用(Bruker Smart APEX CCDⅡ)衍射仪在173(2)K条件下收集衍射数据(SMART程序)。衍射光源经石墨单色化的Mo Kα 射线 (λ=0.071 073 nm), 扫描方式为 φ-ω scan。收集到的衍射数据用SAINT程序还原后再用SADABS程序作吸收校正。结构用SHELXS-97程序由直接法解出[25],并用SHELXL-97程序[25]对非氢原子坐标及其各向异性温度因子进行全矩阵最小二乘法精修至收敛;氢原子坐标由理论计算确定。有关晶体学数据如表1所示,主要键长键角如表2所示。
CCDC:1561437。

表1 配合物1的晶体学数据Table1 Crystallographic data of complex 1
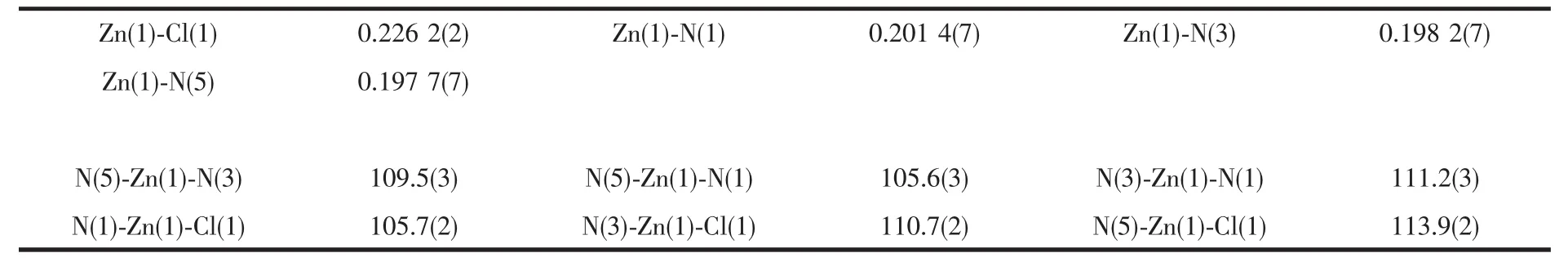
表2 配合物1主要的键长(nm)和键角(°)Table2 Selected bond lengths(nm)and bond angles(°)of complex 1
1.4 细胞培养
将人肝癌细胞(HepG2、Hep3B、Huh7)置于含有体积分数10%胎牛血清、1%青链霉素的DMEM高糖培养基中,在37℃、5%CO2(V/V,下同)条件下培养至第三代对数生长期。
1.5 配合物1的体外抗癌活性测试
1.5.1 对细胞形态的影响
选择对数生长期并且状态良好的HepG2、Hep3B和Huh7细胞分别接种于96孔板,浓度为2×104mL-1,每孔 200 μL。 于 5%CO2,37 ℃培养箱中培养8 h后,细胞贴壁并恢复生长状态后,弃去培养液,并加入1%FBS培养液200 μL。配合物 1用DMSO 溶剂溶解, 稀释至 125.00、62.5、15.63、3.91 μg·mL-1四个加药终浓度,并分别取 1 μL加入到细胞中。在5%CO2,37℃培养箱中培养48 h后,于倒置显微镜下观察细胞形态的变化。
1.5.2 MTT法检测细胞毒性
分别取对数生长期HepG2、Hep3B和Huh7细胞接种于96孔板上,浓度为5×104mL-1,每孔液体为200 μL,即每孔细胞数约为1×104个。将96孔培养板置于5%CO2,37℃培养箱中培养8 h至细胞完全贴壁后,弃去培养液并在每孔加入1%FBS培养液200 μL,饥饿处理2 h。用DMSO将配合物1稀释至浓度 为 250.00、125.00、62.5、31.25、15.63、7.82 μg·mL-1, 实验组每孔滴加药物 1 μL; 药物 ZnCl2、Hppo和阳性对照药物5-氟尿嘧啶(5-FU),每孔滴加1 μL,浓度为 250.00、125.00、62.50、31.25 μg·mL-1(DMSO溶剂)。DMSO最终浓度小于1%,基本不影响细胞活性。另设阴性对照组和溶剂对照组(DMSO)。将加入配合物1的96孔培养板置于5%CO2,37℃培养箱中分别培养 24、48、72 h, 而将加入 ZnCl2、Hppo和5-氟尿嘧啶的96孔培养板培养48 h。离心,弃去上层培养基,各孔加入90 μL不含血清的DMEM培养基与10 μL浓度为5 mg·mL-1的MTT,置于37℃、5%CO2条件下再继续培养4 h。移去上清液,每孔加入 DMSO 150 μL,振荡 5 min,利用酶标仪于490 nm处测定各孔吸光度值,计算细胞存活率,绘制成细胞存活率曲线图,用SPSS计算细胞毒性IC50值。
2 结果与讨论
2.1 配合物[Zn(Hppo)2(ppo)Cl](1)的合成与红外光谱解析
无水ZnCl2与Hppo反应要在加热的条件下反应,温度低会导致ZnCl2析出,影响产率。反应结束后趁热过滤,静置1周左右,即得无色透明块状晶体。晶体产率偏低,溶剂挥发时间增长,会使产率增加,但是晶体多为碎晶。配合物的红外谱图显示,在3 422 cm-1为OH吸收峰,3 339 cm-1为NH吸收峰,在 1 618、1 567、1 501 cm-1,出现苯环及吡唑环ν(C=C)双键吸收峰,1 535 cm-1为 ν(C=N)吸收峰,756、696 cm-1,为单取代苯环 ν(C-H)吸收峰,548 cm-1处出现Zn-N键吸收峰。
2.2 晶体结构描述及表征
图1为配合物1的晶体结构,其主要键长、键角列于表2,其分子内和分子间氢键列于表3。该分子为单核配合物,锌原子为四配位,与3个吡唑环上N原子和1个Cl原子配位形成一个扭曲的四面体构型。其中Zn-N的键长分别为0.201 4(7)、0.198 2(7)和0.197 7(7)nm,Zn-Cl的键长较长,为0.226 2(2)nm;锌与配位原子的键角从105.6(3)°到113.9(2)°。
晶体结构解析表明,N1与锌原子键合,在此种情况下,吡唑环与锌原子可形成3种键合方式,如图2所示:方式Ⅰ中,N1的孤对电子与Zn配位;方式Ⅱ、Ⅲ中,N1失去质子与Zn形成离子键。红外数据中未见C=O吸收峰,说明配合物中不含有方式Ⅲ;1 535 cm-1处的 ν(C=N)吸收峰,较通常 ν(C=N)键(1 690~1 590 cm-1)向短波方向移动,说明N原子参与配位,因此方式Ⅰ为配合物中吡唑结构。3个吡唑配体在与锌形成配合物时,需要失去1个质子达到电荷平衡,羟基氢较氨基氢活泼,因此其中一个羟基失去质子形成负离子。由于3个吡唑环的电子分布情况非常接近,在确定羟基氧负离子时,通过比较连有羟基的C-O键长,C(3)-O(1)为0.134 8(10)nm;C(12)-O(2)为 0.131 6(10)nm;C(21)-O(3)为 0.130 6(10)nm;C(21)-O(3)键长较短,因此把 O(3)确定为去质子,进而得出图1中晶体结构。
在该晶体中,配合物通过3个分子内氢键N(2)-H(2)与 O(3)、N(4)-H(4)与 O(1)、N(6)-H(5)与 O(2)使 3个吡唑配体形成环状构型。分子间是通过O(1)-H(1)与另一分子的 Cl(1)i(Symmetry codes:i1+x,y,z)形成分子间氢键,进而构成一维链状,链与链之间通过分子上的O(2)-H(3)和另一链上的O(3)ii(Symmetry codes:ii1-x,1/2+y,1-z)形成分子间氢键,使配合物形成三维网状结构(图3)。

表3 配合物1的分子内和分子间氢键参数Table3 Intramolecular and intermolecular hydrogen bond parameters of complex 1

图1 配合物1的晶体结构(椭球率30%)Fig.1 Molecular structure of the complex 1 with 30%probability ellipsoids

图2 吡唑与锌的3种键合形式Fig.2 Three binding types of pyrazole and Zn
2.3 配合物1对HepG2、Hep3B和Huh7细胞形态的影响
图4显示为在配合物1作用48 h后,肝癌细胞HepG2、Hep3B和Huh7的细胞形态。通过倒置显微镜观察可见,空白对照组细胞大小均一,胞质颜色透明。随着药物浓度的增加和作用时间的延长,细胞轮廓增强,细胞变圆浮起反差增大,细胞间接触变松,增殖减慢,胞质中颗粒增多。同时,死细胞的数量增多,有的细胞内可见有明显空泡,细胞完整性受到破坏,形成大量细胞碎片。
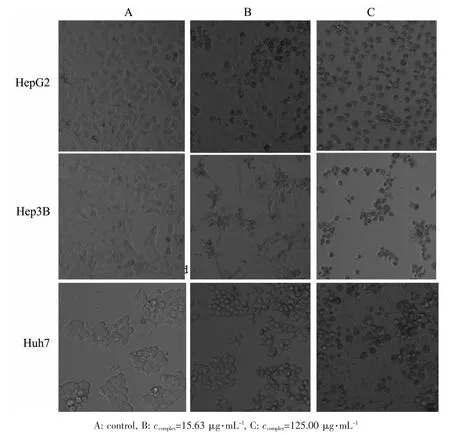
图4 配合物1作用48 h后,倒置显微镜下肝癌细胞HepG2、Hep3B、Huh7的细胞形态(×200)Fig.4 Effect of complex 1 on tumor cell morphology of HepG2,Hep3B,Huh7 cell lines after 48 h investigated by inverted phase-contrast microscopy(×200)
2.4 配合物1对HepG2、Hep3B和Huh7的细胞毒性
实验采用 MTT法检测金属配合物 1、ZnCl2、Hppo和5-FU对HepG2、Hep3B和Huh7的细胞毒性。在加药24、48、72 h后,不同药物对肝癌细胞HepG2、Hep3B和Huh7造成了不同程度的抑制作用。配合物1具有较强的抑制效果,图5显示了配合物1对HepG2、Hep3B和 Huh7细胞, 在 24、48、72 h的抑制情况。表4总结了药品的IC50,其中对Hep3B抑制效果最好,48 h时的IC50值为9.88 μg·mL-1, 对 HepG2 细胞的 IC50值为 20.73 μg·mL-1,对Huh7 细胞的抑制作用最弱,IC50值为 52.16 μg·mL-1。金属配合物1对肝癌细胞的抑制均呈剂量依赖,即随药物浓度的增加细胞的抑制效果增强。在给药时间为24~48 h时,抑制率随着作用时间的增强而增强,呈正相关。

图5 配合物1对肿瘤细胞HepG2、Hep3B和Huh7的抑制率曲线(n=3)Fig.5 Inhibition rate of complex 1 against tumor cell lines HepG2,Hep3B and Huh7(n=3)with the increasing of concentration
通过对比配合物 1与 ZnCl2、Hppo和5-FU对HepG2、Hep3B和Huh7的抑制作用发现(图6),在样品浓度62.50 μg·mL-1下,吡唑配体和金属盐对细胞抑制效果较弱,金属配合物1对Hep3B和HepG2抑制效果与5-氟尿嘧啶的抑制效果相当,对Hep3B细胞的作用效果甚至强于阳性对照药,对Huh7的抑制效果较弱,与上面实验相符。在已报道的吡唑类金属配合物的抗肿瘤作用机制研究中,吡唑配体多数以非共价键结合方式与DNA相互作用[14]。配合物1中含有苯环和吡唑环,可以通过插入方式嵌入DNA,这有可能是配合物的抗肿瘤活性原因之一。

表4 配合物1对Hep3B、HepG2和Huh7细胞的IC50值(n=3)Table4 IC50of complex 1 on tumor cell Hep3B,HepG2 and Huh7(n=3)μg·mL-1
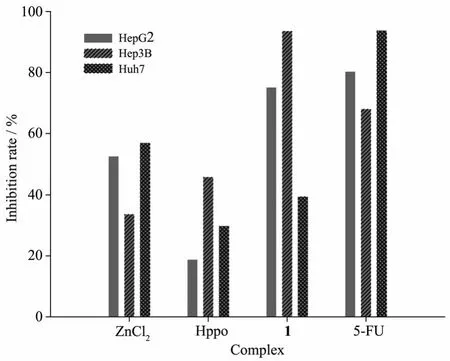
图6 化合物对细胞Hep3B、HepG2和Huh7的抑制率(n=3)Fig.6 Inhibition rate of the compounds against HepG2,Hep3B and Huh7(n=3)
3 结 论
合成了新型金属配合物[Zn(Hppo)2(ppo)Cl]并表征了其晶体结构。Znギ为四面体配位,配体5-苯基-3-羟基吡唑(Hppo)通过吡唑环上的N原子与Zn配位,分子上的OH和NH通过形成分子内和分子间氢键稳定配合物结构。体外细胞实验显示配合物1对肝癌细胞HepG2,Hep3B和Huh7具有增值抑制作用,其中对Hep3B的抑制效果最好,强于阳性对照5-氟尿嘧啶。ZnCl2与Hppo对3种肝癌细胞的抑制效果较弱,在形成配合物后生物活性增强。
[1]Mariappan G,Saha B P,Sutharson L,et al.Saudi Pharm.J.,2011,19(2):115-122
[2]Bekhit A A,Abdel-Aziem T.Bioorg.Med.Chem.,2004,12(8):1935-1945
[3]Hashimoto H,Imamura K,Haruta J,et al.J.Med.Chem.,2002,45(7):1511-1517
[4]Peng X M,Cai G X,Zhou C H.Curr.Top.Med.Chem.,2013,13(16):1963-2010
[5]Keter F K,Darkwa J.BioMetals,2012,25(1):9-21
[6]Marcheetti F,Pettinari C,Pettinari R.Coord.Chem.Rev.,2005,249(24):2909-2945
[7]Casas J S,García-Tasende M S,Sánchez A,et al.Coord.Chem.Rev.,2007,251(11/12):1561-1589
[8]Mariappan G,Sha B P,Sutharson L,et al.J.Pharm.Res.,2010,3(12):2856-2859
[9]HUANG Juan(黄娟),CUI Zi-Ning(崔紫宁),LI Ying(李映),et al.Chin.Org.Chem.(有机化学),2008,28(4):598-604
[10]Caruso F,Rossi M,Tanski J,et al.J.Med.Chem.,2000,43(20):3665-3670
[11]Budzisz E,Malecka M,Keppler B K,et al.Eur.J.Inorg.Chem.,2007(23):3728-3735
[12]HE Qi-Zhuang(何其庄),MA Shu-Zhi(马树芝),XU Dong-Fang(许东芳).Chinese J.Inorg.Chem.(无机化学学报),2007,23(10):1723-1728
[13]Marzano C,Pellei M,Tisato F,et al.Anticancer Agents Med.Chem.,2009,9(2):185-211
[14]ZHANG Si-Qi(张思琪),WANG Lu(王路),WANG Zhen-Yu(王振宇).Chem.Res.Appl.(化学研究与应用),2014,26(6):777-784
[15]LIU Ya-Nan(刘亚楠),YANG Fang(杨芳),MEI Wen-Jie(梅文杰),et al.Chem.J.Chinese Universities(高等学校化学学报),2010,31(3):435-441
[16]Jiang J,Tang X L,Dou W,et al.J.Inorg.Biochem.,2010,104(5):583-591
[17]Li M X,Chen C L,Zhang D,et al.Eur.J.Med.Chem.,2010,45(7):3169-3177
[18]Zhang H,Liu C S,Bu X H,et al.J.Inorg.Biochem.,2005,99(5):1119-1125
[19]QIU Xiao-Yang(仇晓阳),LIU Qi-Feng(刘起峰),ZHANG Ping(张平),et al.Chinese J.Inorg.Chem.(无机化学学报),2012,28(2):362-366
[20]Kovala-Demertzi D,Yadav P N,Wiecek J,et al.J.Inorg.Biochem.,2006,100(9):1558-1567
[21]Stanojkovic T P,Kovala-Demertzi D,Primikyri A,et al.J.Inorg.Biochem.,2010,104(4):467-476
[22]Lopes E O,Oliveira C G,Silva P B,et al.Int.J.Mol.Sci.,2016,17(5):781-795
[23]Aromí A,Bell A R,Helliwell M,et al.Chem.Eur.J.,2003,9:3024-3032
[24]Zimmermann D,Krogsgaard-Larsen P,Ehrhardt J D,et al.Tetrahedron,1998,54(32):9393-9400
[25]Sheldrick G M.SHELXL-97,Program for the Solution and the Refinement of Crystal Structure,University of Göttingen,Germany,1997.
Synthesis,Crystal Structure and Antitumor Activity of Pyrazole Zinc Complex
LI Jing*WU Chun-Yang ZHANG Ying SHEN Gui-Nan JIN Cheng-Hao
(College of Life Science and Biotechnology,Heilongjiang Bayi Agriculture University,Daqing,Heilongjiang 163319,China)
A zinc complex[Zn(Hppo)2(ppo)Cl](1)(Hppo=3-benzyl-3-pyrazolin-5-one)was synthesized by solvent evaporation and characterized by elemental analysis,FTIR and X-ray diffraction.The complex crystallizes in monoclinic system,space group P21.Cell morphology and in vitro toxicity effects against cancer cell lines HepG2,Hep3B and Huh7 of the complex were investigated.The bioassay results show that this zinc complex has distinct antitumor effects.CCDC:1561437.
pyrazole;antitumor activity;metal complex;crystal structure
O614.24+1
A
1001-4861(2018)01-0135-07
10.11862/CJIC.2018.016
2017-03-30。收修改稿日期:2017-11-10。
国家自然科学基金(No.31400660)、黑龙江八一农垦大学青年创新人才基金(No.CXRC2016-13)和黑龙江省自然科学基金(No.QC2016012)资助项目。
*通信联系人。 E-mail:lijingroea@sina.com
